Dernière mise à jour le 01/06/2026
DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Faible | Avis du 03/05/2017 | Renouvellement d'inscription (CT) | Le service médical rendu par les spécialités DEBRIDAT reste faible dans les indications : traitement symptomatique des douleurs liées aux troubles fonctionnels du tube digestif, traitement symptomatique des douleurs, troubles du transit et inconfort intestinal liés aux troubles fonctionnels intestinaux chez l’adulte et chez l’enfant de plus de 2 ans. |
| Insuffisant | Avis du 03/05/2017 | Renouvellement d'inscription (CT) | Le service médical rendu par les spécialités DEBRIDAT reste insuffisant dans l‘indication du traitement symptomatique des douleurs liées aux troubles fonctionnels des voies biliaires. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 26/08/2022
DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Trimébutine........................................................................................................................... 0,7870 g
Pour 100 g de granulés pour suspension buvable.
76,25 g de granulés correspondent à 125 ml de suspension reconstituée.
Excipients à effet notoire : chaque flacon (de 76,25 g de granulés) contient 1,372 mg de jaune orangé S et 74,072 g de saccharose.
Pour la liste complète des excipients, voir rubrique 6.1.
Granulés pour suspension buvable.
4.1. Indications thérapeutiques
Traitement symptomatique des douleurs, des troubles du transit et de l’inconfort intestinal liés aux troubles fonctionnels intestinaux.
4.2. Posologie et mode d'administration
DEBRIDAT ENFANT est indiqué chez l'enfant entre 2 et 5 ans.
Au-delà de 5 ans, il convient d’utiliser des formes pharmaceutiques plus adaptées.
Reconstituer les granulés pour suspension buvable par addition d’eau non gazeuse jusqu’au repère figurant sur le flacon et agiter afin de bien mélanger la préparation.
La posologie quotidienne totale habituelle est de 1 mL/kg/jour administrée en 3 prises. Par exemple, pour un enfant de 15 kg : remplir la seringue jusqu’à la graduation de 15 kg, 3 fois par jour (matin, midi et soir).
La dose est basée sur le poids corporel et doit être mesurée à l'aide de la seringue de mesure fournie qui est graduée en kg de poids corporel par prise.
Pour les enfants en bas âge, il est recommandé d'ajouter le contenu de la seringue graduée à un biberon d'eau ou de lait et de l'agiter, et de le donner immédiatement à boire à l’enfant.
Il est recommandé d’agiter le flacon avant chaque utilisation.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Enfant de moins de 2 ans.
4.4. Mises en garde spéciales et précautions d'emploi
Excipients
Ce médicament contient un agent colorant azoïque (jaune orangé S) et peut provoquer des réactions allergiques.
Ce médicament contient du saccharose. Les patients présentant une intolérance au fructose, un syndrome de malabsorption du glucose et du galactose ou un déficit en sucrase/isomaltase (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Ce médicament contient environ 0,6 g de saccharose par ml, soit environ 0,20 g par graduation (exemple: la graduation 15 kg contient 3 g de saccharose). Ceci est à prendre en compte pour les patients atteints de diabète sucré.
Ce médicament peut être nocif pour les dents s’il doit être pris de manière prolongée.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Sans objet.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études chez l'animal n'ont pas mis en évidence d'effet tératogène.
Il n'existe pas actuellement de données suffisamment pertinentes, pour évaluer un éventuel effet malformatif ou fœtotoxique de la trimébutine lorsqu'elle est administrée pendant la grossesse.
En conséquence, par mesure de précaution, il est préférable de ne pas utiliser la trimébutine au cours du premier trimestre de la grossesse. En l'absence d'effet néfaste attendu pour la mère ou l'enfant, l'utilisation de la trimébutine au cours des 2ème et 3ème trimestres de la grossesse ne doit être envisagée que si nécessaire.
Allaitement
Le passage dans le lait maternel de la trimébutine n’est pas connu.
Par mesure de précaution, il est préférable d’éviter d’utiliser la trimébutine pendant l’allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Sans objet.
La liste ci-dessous des effets indésirables est issue de l’expérience des essais cliniques et des données rapportées depuis la mise sur le marché.
Selon le système de classification par organe, les effets indésirables sont listés ci-dessous par ordre de fréquence en utilisant les catégories suivantes : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1,000 à < 1/100) ; rare (≥ 1/10,000 à < 1/1,000) ; très rare (< 1/10,000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Affections du système immunitaire
Fréquence indéterminée : réactions d’hypersensibilité (prurit, urticaire, œdème de Quincke et exceptionnellement choc anaphylactique).
Affections de la peau et du tissu sous-cutané
Peu fréquent : rash.
Fréquence indéterminée : éruption maculopapuleuse généralisée, érythèmes, réactions eczématiformes et exceptionnellement réactions cutanées sévères comprenant des cas de pustulose exanthématique aiguë généralisée (PEAG), érythème polymorphe, toxidermie fébrile.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
En cas de surdosage, des troubles cardiaques à type de bradycardie, allongement de l’intervalle QTc, ou tachycardie et des troubles neurologiques à type de somnolence, convulsion et coma ont pu être observés. Une surveillance en milieu spécialisé s’impose et un traitement symptomatique sera à mettre en œuvre.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : ANTISPASMODIQUE MUSCULOTROPE, code ATC : A03AA05.
(A : appareil digestif et métabolisme)
Les effets de la trimébutine s'exercent au niveau du tube digestif sur la motricité intestinale.
La trimébutine a des propriétés d'agoniste enképhalinergique. Elle stimule la motricité intestinale en déclenchant des ondes de phase III propagées du complexe moteur migrant et en l'inhibant lors de stimulation préalable (chez l'animal).
In vitro, elle agit par blocage des canaux sodiques (IC50 = 8.4 μM) et inhibe la libération d'un médiateur de la nociception (le glutamate).
Chez le rat, elle inhibe la réaction de l'animal à la distension rectale et colique dans différents modèles expérimentaux.
5.2. Propriétés pharmacocinétiques
Absorption
Le taux sanguin maximum de trimébutine après l’administration orale de comprimés a été obtenu après 1 à 2 heures.
Élimination
L’élimination de la trimébutine après l’administration orale de comprimés a été principalement urinaire et rapide: 70 % en moyenne en 24 heures.
5.3. Données de sécurité préclinique
Les études de toxicité à doses répétées jusqu’à 6 mois par voie orale avec la trimébutine n’ont pas montré d’effet toxicologique délétère chez le rat et le chien. Des études de génotoxicité (test d’Ames in vitro, aberration chromosomique et le test du micronoyau in vivo) n’ont pas montré d’effet mutagène ou clastogène de la trimébutine. La trimébutine n’a pas d’effet sur le développement et la fertilité des rats mâles et femelles. Les études de la reproduction et du développement sur la trimébutine n'ont pas mis en évidence d’effet tératogène chez le rat et le lapin. Les études de carcinogénicité sur la trimébutine n’ont pas été réalisées.
Polysorbate 80, arôme naturel orange en poudre*, jaune orangé S (E 110), saccharose.
*Composition de l’arôme naturel orange en poudre : huile essentielle d’orange, gomme arabique.
Sans objet.
3 ans.
Après reconstitution, la suspension buvable ne doit pas être conservée plus de 4 semaines.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25° C.
Pour les conditions de conservation du médicament après reconstitution, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
76,25 g en flacon (verre brun) avec seringue (PE/Polystyrène) graduée en kg de poids corporel par prise (une graduation de 15 kg correspond à 5 ml de solution reconstituée).
6.6. Précautions particulières d’élimination et de manipulation
Pas d'exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
PFIZER HOLDING FRANCE
23-25, AVENUE DU DOCTEUR LANNELONGUE
75014 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 341 048 3 1 : 76,25 g en flacon (verre brun) avec seringue graduée (PE/Polystyrène).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste II
ANSM - Mis à jour le : 26/08/2022
DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon
Trimébutine
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon ?
3. Comment prendre DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : ANTISPASMODIQUE MUSCULOTROPE - code ATC : A03AA05.
Ce médicament est indiqué chez l’enfant à partir de 2 ans dans les douleurs spasmodiques de l’intestin.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon ?
Ne prenez jamais DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon :
· si vous êtes allergique à la trimébutine ou à l'un des autres composants contenus
dans ce médicament, mentionnés dans la rubrique 6.
Ne donnez pas ce médicament à un enfant de moins de 2 ans.
EN CAS DE DOUTE, IL EST INDISPENSABLE DE DEMANDER L'AVIS DE VOTRE MEDECIN OU DE VOTRE PHARMACIEN
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon.
Enfants et adolescents
Sans objet.
Autres médicaments et DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris un médicament obtenu sans ordonnance.
DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Ce médicament ne sera utilisé pendant la grossesse que sur les conseils de votre médecin. Si vous découvrez que vous êtes enceinte pendant le traitement, consultez votre médecin car lui seul peut juger de la nécessité de le poursuivre.
Il est préférable de ne pas prendre ce médicament si vous allaitez.
Conduite de véhicules et utilisation de machines
Sans objet.
DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon contient du saccharose et du jaune orangé S.
Ce médicament contient un agent colorant azoïque (jaune orangé S) et peut provoquer des réactions allergiques.
Ce médicament contient du saccharose. Si votre médecin vous a informé(e) que vous avez une intolérance à certains sucres, contactez votre médecin avant de prendre ce médicament.
Ce médicament contient 0,20 g de saccharose par graduation de la seringue. Ceci est à prendre en compte pour les patients atteints de diabète sucré.
Ce médicament peut être nocif pour les dents.
3. COMMENT PRENDRE DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon ?
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Posologie
Ce modèle est adapté à l’enfant.
DEBRIDAT ENFANT est indiqué chez l'enfant entre 2 et 5 ans.
Au-delà de 5 ans, il convient d’utiliser des formes pharmaceutiques plus adaptées.
La posologie quotidienne totale habituelle est de 1 mL/kg/jour administrée en 3 prises. Par exemple, pour un enfant de 15 kg : remplir la seringue jusqu’à la graduation de 15 kg, 3 fois par jour (matin, midi et soir).
La dose est basée sur le poids corporel et doit être mesurée à l'aide de la seringue de mesure fournie qui est graduée en kg de poids corporel par prise. Des exemples ont été données dans le tableau ci-dessous.
Jusqu’à 15kg : remplir la seringue jusqu’à la graduation indiquant le poids de l’enfant.
Entre 15kg et 30kg : remplir une première fois la seringue jusqu’à la graduation 15kg, puis une deuxième fois jusqu’à atteindre un total égal au poids de l’enfant.
Mesure de la dose par prise chez les enfants entre 8 kg et 30 kg :
|
Poids de l’enfant |
Remplir la seringue jusqu’à la graduation de : |
Puis remplir la seringue, la deuxième fois, jusqu’à la graduation de : |
|
8 kg |
8 kg |
Non Applicable |
|
9 kg |
9 kg |
Non Applicable |
|
10 kg |
10 kg |
Non Applicable |
|
11 kg |
11 kg |
Non Applicable |
|
12 kg |
12 kg |
Non Applicable |
|
13 kg |
13 kg |
Non Applicable |
|
14 kg |
14 kg |
Non Applicable |
|
15 kg |
15 kg |
Non Applicable |
|
16 kg |
15 kg |
1 kg |
|
17 kg |
15 kg |
2 kg |
|
18 kg |
15 kg |
3 kg |
|
19kg |
15 kg |
4 kg |
|
20 kg |
15 kg |
5 kg |
|
21 kg |
15 kg |
6 kg |
|
22 kg |
15 kg |
7 kg |
|
23 kg |
15 kg |
8 kg |
|
24 kg |
15 kg |
9 kg |
|
25 kg |
15 kg |
10 kg |
|
26 kg |
15 kg |
11 kg |
|
27 kg |
15 kg |
12 kg |
|
28 kg |
15 kg |
13 kg |
|
29 kg |
15 kg |
14 kg |
|
30 kg |
15 kg |
15 kg |
Mode et voie d'administration
Voie orale.
Préparation de la suspension buvable :
· introduire jusqu’au trait de l’eau minérale non gazeuse,
· refermer le flacon et bien agiter pour obtenir un liquide homogène,
· sortir la seringue graduée,
· visser la seringue graduée sur le flacon.
Mode d’emploi :
· agiter le flacon avant chaque utilisation,
· tirer sur le piston jusqu’à la graduation correspondant à la dose requise de la prise, en maintenant la collerette,
· prendre la seringue par la collerette et la tirer en traversant le bouchon sans toucher au piston,
· verser le contenu de la seringue graduée dans un biberon, pour les enfants en bas âge,
· maintenir l’enfant en position debout pendant l’administration,
· introduire la seringue dans la bouche de l’enfant sans l’enfoncer, en la dirigeant sur la face interne de la joue, et administrer la totalité de la suspension en appuyant doucement et progressivement sur le piston,
· rincer soigneusement la seringue graduée à l’eau claire,
· remettre la seringue graduée dans le flacon à travers le bouchon.
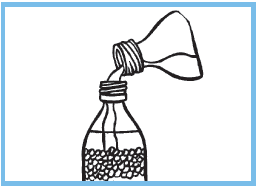
1.Introduire jusqu’au trait de l’eau minérale non gazeuse,
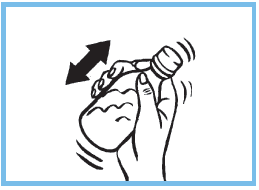
2. Refermer le flacon et bien agiter pour obtenir un liquide homogène
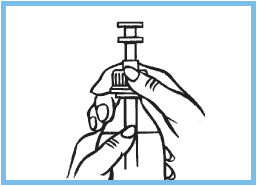
3. Sortir la seringue graduée. Visser la seringue graduée sur le flacon.
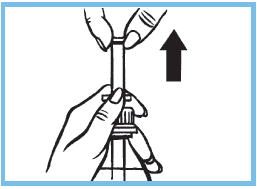
4. Agiter le flacon avant chaque utilisation. Tirer sur le piston jusqu’à la graduation correspondant au poids de l’enfant, en maintenant la collerette. Voir le tableau ci-dessus concernant la quantité à prendre pour chaque prise.
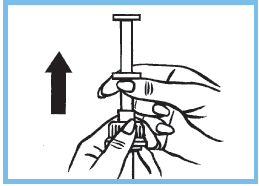
5. Prendre la seringue par la collerette et la tirer en traversant le bouchon sans toucher au piston. Verser le contenu de la seringue graduée dans un biberon, pour les enfants en bas âge, et le faire boire immédiatement.
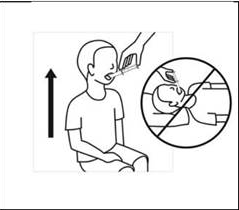
6. Maintenir l’enfant en position debout pendant l’administration. Introduire la seringue dans la bouche de l’enfant sans l’enfoncer, en la dirigeant sur la face interne de la joue, et administrer la totalité de la suspension en appuyant doucement et progressivement sur le piston
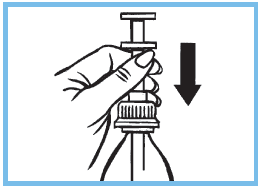
7. Rincer soigneusement la seringue graduée à l’eau claire. Remettre la seringue graduée dans le flacon à travers le bouchon.
Fréquence et moment auxquels le médicament doit être administré
1 prise à renouveler 3 fois par jour.
Si vous avez pris plus de DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon que vous n’auriez dû
Consultez immédiatement votre médecin.
Vous pouvez présenter les symptômes suivants : problèmes cardiaques, somnolence, convulsions, perte de conscience (coma).
Une surveillance en milieu spécialisé pourra être nécessaire et un traitement symptomatique administré.
Si vous oubliez de prendre DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon
Sans objet.
Si vous arrêtez de prendre DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Les effets indésirables suivants peuvent survenir :
Peu fréquent (peut affecter jusqu’à une personne sur 100) :
· Eruption cutanée.
Fréquence indéterminée (les informations disponibles ne permettent pas d’évaluer la fréquence) :
· Réactions allergiques : démangeaisons, urticaire, œdème de la face (œdème de Quincke), choc anaphylactique.
· Manifestations cutanées : éruption en relief (macules, papules), érythème, eczéma (dermite de contact).
· Réactions cutanées sévères : rougeur se généralisant à tout le corps avec des pustules et accompagnée de fièvre (pustulose exanthématique aiguë généralisée), rougeur de la peau accompagnée de fièvre (érythème polymorphe, toxidermie).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le conditionnement extérieur. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 25 °C.
Après reconstitution, la suspension buvable ne doit pas être conservée plus de 4 semaines.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient DEBRIDAT ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon
· La substance active est :
Trimébutine................................................................................................................... 0,7870 g
Pour 100 g de granulés pour suspension buvable
· Les autres composants sont :
Polysorbate 80, arôme naturel orange (huile essentielle d’orange, gomme arabique), jaune orangé S (E 110), saccharose (voir rubrique 2 « DEBRIDAT, ENFANT 4,8 mg/ml, granulés pour suspension buvable en flacon contient du saccharose et du jaune orangé S »).
Ce médicament se présente sous forme de granulés pour suspension buvable. Boîte de 1 flacon de 76,25 g de granulés et une seringue graduée en kg de poids corporel par prise (une graduation de 15 kg correspond à 5 ml de solution reconstituée).
Titulaire de l’autorisation de mise sur le marché
PFIZER HOLDING FRANCE
23-25, AVENUE DU DOCTEUR LANNELONGUE
75014 PARIS
Exploitant de l’autorisation de mise sur le marché
PFIZER
23-25, AVENUE DU DOCTEUR LANNELONGUE
75014 PARIS
FARMEA
10, RUE BOUCHE THOMAS
ZAC d’Orgemont,
49000 ANGERS
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Sans objet.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).