Dernière mise à jour le 28/04/2026
PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable
Indications thérapeutiques
PAROXETINE VIATRIS est un traitement destiné aux adultes souffrant de dépression et/ou de troubles anxieux.
Les troubles anxieux dans lesquels PAROXETINE VIATRIS peut être prescrit sont les suivants :
· troubles obsessionnels compulsifs (pensées répétitives, obsessionnelles avec comportement incontrôlable),
· trouble panique (attaques de panique, y compris celles causées par la peur des lieux publics, l'agoraphobie)
· trouble anxiété sociale (peur ou rejet de situations où vous devez être en société)
· état de stress post-traumatique (anxiété causée par un événement traumatique)
· anxiété généralisée.
PAROXETINE VIATRIS appartient à la classe de médicaments appelés Inhibiteurs Sélectifs de la Recapture de la Sérotonine (ISRS).
Le mécanisme d'action de PAROXETINE VIATRIS et des autres ISRS n'est pas complètement connu, mais ils augmenteraient le taux de sérotonine dans le cerveau.
Bien traiter votre dépression ou votre trouble anxieux est important pour vous aider à vous sentir mieux.
Présentations
> 1 flacon(s) polyéthylène de 14 comprimé(s)
Code CIP : 359 987-1 ou 34009 359 987 1 2
Déclaration de commercialisation : 28/08/2013
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 2,20 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 3,22 €
- Taux de remboursement :65%
> plaquette(s) aluminium de 14 comprimé(s)
Code CIP : 374 173-1 ou 34009 374 173 1 0
Déclaration de commercialisation : 13/09/2006
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 2,20 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 3,22 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 22/02/2017 | Inscription (CT) | Le service médical rendu par PAROXETINE MYLAN 20 mg est important dans les indications : « Episode dépressif majeur », « Troubles Obsessionnels Compulsifs », « Trouble Panique avec ou sans agoraphobie », « Trouble Anxiété Généralisée », « Etat de stress post-traumatique ». |
| Modéré | Avis du 22/02/2017 | Inscription (CT) | Le service médical rendu par PAROXETINE MYLAN 20 mg est modéré dans l’indication « Trouble Anxiété Sociale / Phobie sociale ». |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 22/02/2017 | Inscription (CT) | Cette spécialité est un générique qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport au princeps DEROXAT 20 mg. |
ANSM - Mis à jour le : 24/07/2025
PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Sous forme de chlorhydrate de paroxétine anhydre
Pour un comprimé pelliculé sécable.
Pour la liste complète des excipients, voir rubrique 6.1.
4.1. Indications thérapeutiques
· Episode dépressif majeur.
· Troubles Obsessionnels Compulsifs.
· Trouble Panique avec ou sans agoraphobie.
· Trouble Anxiété Sociale / Phobie sociale.
· Trouble Anxiété Généralisée.
· Etat de stress post-traumatique.
4.2. Posologie et mode d'administration
Episode dépressif majeur
La posologie recommandée est de 20 mg par jour.
En général, l'amélioration du patient débute après une semaine de traitement mais peut ne devenir manifeste qu'à partir de la deuxième semaine.
Comme avec tous les médicaments antidépresseurs, la posologie doit être revue et ajustée si nécessaire au cours des 3 à 4 semaines suivant le début du traitement et par la suite si cela est cliniquement justifié.
Chez certains patients présentant une réponse insuffisante sous 20 mg, la posologie peut être augmentée graduellement par palier de 10 mg en fonction de la réponse thérapeutique, jusqu'à un maximum de 50 mg par jour.
Les patients souffrant de dépression doivent être traités pendant une période suffisante d'au moins 6 mois afin d'assurer la disparition des symptômes.
Troubles obsessionnels compulsifs (TOC)
La posologie recommandée est de 40 mg par jour. Le traitement sera débuté à la dose de 20 mg par jour, qui pourra être augmentée progressivement par palier de 10 mg jusqu'à la dose recommandée.
En cas de réponse insuffisante après plusieurs semaines de traitement à la dose recommandée, certains patients peuvent tirer bénéfice d'une augmentation progressive de dose, jusqu'à un maximum de 60 mg par jour.
Les patients souffrant de TOC doivent être traités pendant une période suffisante afin d'assurer la disparition des symptômes. Cette période peut durer plusieurs mois voire même plus longtemps (voir rubrique 5.1).
Trouble panique
La posologie recommandée est de 40 mg par jour. Le traitement sera débuté à la dose de 10 mg par jour, qui pourra être augmentée progressivement par palier de 10 mg en fonction de la réponse thérapeutique jusqu'à la dose recommandée.
Une faible dose initiale est recommandée afin de minimiser l'aggravation potentielle des symptômes du trouble panique, pouvant survenir en début de traitement. En cas de réponse insuffisante après plusieurs semaines de traitement à la dose recommandée, certains patients peuvent tirer bénéfice d'une augmentation progressive de dose, jusqu'à un maximum de 60 mg par jour.
Les patients atteints de trouble panique doivent être traités pendant une période suffisante afin d'assurer la disparition des symptômes. Cette période peut durer plusieurs mois voire même plus longtemps (voir rubrique 5.1).
Trouble anxiété sociale/phobie sociale
La posologie recommandée est de 20 mg par jour. En cas de réponse insuffisante après plusieurs semaines de traitement à la dose recommandée, certains patients peuvent tirer bénéfice d'une augmentation progressive de dose par palier de 10 mg, jusqu'à un maximum de 50 mg par jour.
L'utilisation à long terme doit être régulièrement évaluée (voir rubrique 5.1).
Trouble anxiété généralisée
La posologie recommandée est de 20 mg par jour. En cas de réponse insuffisante après plusieurs semaines de traitement à la dose recommandée, certains patients peuvent tirer bénéfice d'une augmentation progressive de dose par palier de 10 mg, jusqu'à un maximum de 50 mg par jour.
L'utilisation à long terme doit être régulièrement évaluée (voir rubrique 5.1).
Etat de stress post-traumatique
La posologie recommandée est de 20 mg par jour. En cas de réponse insuffisante après plusieurs semaines de traitement à la dose recommandée, certains patients peuvent tirer bénéfice d'une augmentation progressive de dose par palier de 10 mg par semaine, jusqu'à un maximum de 50 mg par jour. L'utilisation à long terme doit être régulièrement évaluée (voir rubrique 5.1).
Symptômes de sevrage observes lors de l'arrêt de la paroxétine
Un arrêt brutal du traitement doit être évité (voir rubriques 4.4 et 4.8).
Le schéma utilisé au cours des essais cliniques comportait une interruption progressive de traitement avec diminution de la dose journalière par palier de 10 mg par semaine.
La survenue de symptômes gênants lors de la diminution de la dose ou à l'arrêt du traitement pourra nécessiter la reprise de la dose précédemment prescrite. Le médecin pourra ensuite poursuivre la diminution de la dose à un rythme plus progressif.
Enfants et adolescents (7-17 ans)
La paroxétine est déconseillée chez l’enfant et l’adolescent, des études cliniques contrôlées ayant montré que la paroxétine était associée à un risque accru de comportement suicidaire et d’hostilité. De plus, l’efficacité de la paroxétine n’a pas été suffisamment démontrée dans ces essais (voir rubriques 4.4 et 4.8).
Enfants âgés de moins de 7 ans
L’utilisation de la paroxétine n’a pas été étudiée chez l’enfant de moins de 7 ans. La paroxétine est déconseillée tant que son efficacité et sa sécurité d’emploi n’ont pas été démontrées dans cette tranche d’âge.
Population âgée
Une augmentation des concentrations plasmatiques est observée chez les sujets âgés mais elles demeurent cependant dans les limites de celles observées chez les patients plus jeunes. La posologie initiale est la même que chez l'adulte. Une augmentation de dose pourra être utile chez certains patients, mais la dose maximale ne devra pas excéder 40 mg par jour.
Insuffisance hépatique ou rénale :
Une augmentation des concentrations plasmatiques de la paroxétine est observée chez l'insuffisant rénal sévère (clairance de la créatinine < 30 ml/min) ainsi que chez l'insuffisant hépatique. La posologie recommandée la plus faible ne devrait donc pas être dépassée chez ces patients.
Mode d’administration
Il est recommandé d'administrer la paroxétine en une prise journalière, le matin au cours du petit déjeuner.
Les comprimés doivent être avalés plutôt que croqués.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1
La paroxétine est contre-indiquée en association aux Inhibiteurs de la Monoamine Oxydase (IMAO). Dans des circonstances exceptionnelles, le linézolide (un antibiotique IMAO non sélectif réversible) peut être utilisé en association avec la paroxétine à condition d’être en mesure d’assurer une surveillance étroite permettant de détecter les symptômes évocateurs d’un syndrome sérotoninergique et un suivi de la pression artérielle (voir rubrique 4.5).
Le traitement avec la paroxétine pourra être instauré :
· 2 semaines après l’arrêt d’un traitement par un IMAO irréversible, ou
· au moins 24 heures après l’arrêt d’un IMAO réversible (ex : moclobémide, linézolide, chlorure de méthylthioninium (bleu de méthylène ; agent de marquage préopératoire qui est un IMAO non sélectif réversible).
Respecter un délai d’au moins une semaine entre l’arrêt de la paroxétine et le début du traitement par un IMAO.
La paroxétine est contre-indiquée en association avec la thioridazine ou le pimozide (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
Population pédiatrique
L’utilisation de PAROXETINE VIATRIS est déconseillée chez les enfants et adolescents de moins de 18 ans. Des comportements de type suicidaire (tentatives de suicides et idées suicidaires) et de type hostile (principalement agressivité, comportement d’opposition et colère) ont été plus fréquemment observés aux cours des études cliniques chez les enfants et adolescents traités par antidépresseurs par rapport à ceux traités par placebo.
Si, en cas de nécessité clinique, la décision de traiter est néanmoins prise, le patient devra faire l’objet d’une surveillance attentive pour détecter l’apparition de symptômes suicidaires. De plus, on ne dispose d’aucune donnée de tolérance à long terme chez l’enfant et l’adolescent concernant la croissance, la maturation et le développement cognitif et comportemental.
Suicide/idées suicidaires ou aggravation clinique
La dépression est associée à un risque accru d’idées suicidaires, d’auto-agression et de suicide (comportement de type suicidaire). Ce risque persiste jusqu’à obtention d’une rémission significative.
L’amélioration clinique pouvant ne pas survenir avant plusieurs semaines de traitement, les patients doivent être étroitement surveillés jusqu’à obtention de cette amélioration. L’expérience clinique montre que le risque suicidaire peut augmenter en tout début de rétablissement.
Les autres troubles psychiatriques dans lesquels la paroxétine est prescrite peuvent également être associés à un risque accru de comportement suicidaire. De plus, ces troubles peuvent être associés à un épisode dépressif majeur.
Les mêmes précautions d’emploi que celles prises pour les patients souffrant d’épisodes dépressifs majeurs devront donc être appliquées aux patients présentant d’autres troubles psychiatriques.
Les patients ayant des antécédents de comportement de type suicidaire ou ceux exprimant des idées suicidaires significatives avant de débuter le traitement présentent un risque plus élevé de survenue d’idées suicidaires ou de comportements de type suicidaire, et doivent faire l’objet d’une surveillance étroite pendant le traitement.
Une méta-analyse d’essais cliniques contrôlés versus placebo sur l’utilisation d’antidépresseurs chez l’adulte présentant des troubles psychiatriques a montré une augmentation du risque de comportement de type suicidaire chez les patients de moins de 25 ans traités par antidépresseurs par rapport à ceux recevant un placebo (voir rubrique 5.1).
Une surveillance étroite des patients, et en particulier de ceux à haut risque, devra accompagner le traitement médicamenteux, particulièrement au début du traitement et lors des changements de dose.
Les patients (et leur entourage) doivent être avertis de la nécessité de surveiller la survenue d’une aggravation clinique, l’apparition d’idées/comportements suicidaires et tout changement anormal du comportement et de prendre immédiatement un avis médical si ces symptômes survenaient.
Akathisie/agitation psychomotrice
L’utilisation de la paroxétine a été associée à l’apparition d’akathisie, caractérisée par une sensation intérieure d’impatience et d’agitation psychomotrice, telle qu’une impossibilité de rester assis ou debout tranquillement, associée en général à un sentiment de désarroi. Ces symptômes surviennent plutôt dans les premières semaines de traitement. Chez les patients développant ces symptômes, une augmentation de posologie peut être préjudiciable.
Syndrome sérotoninergique/syndrome malin des neuroleptiques
Dans de rares cas, un syndrome sérotoninergique ou un tableau évocateur de syndrome malin des neuroleptiques peuvent survenir lors du traitement par la paroxétine, en particulier lorsque celle-ci est associée à des médicaments sérotoninergiques et/ou des neuroleptiques,
Ces syndromes pouvant menacer le pronostic vital, le traitement par la paroxétine devra être arrêté si de tels effets surviennent (caractérisés par un ensemble de symptômes tels qu’hyperthermie, rigidité, myoclonies, dysautonomie accompagnée de possibles fluctuations rapides des constantes vitales, modification de l’état psychique incluant confusion, irritabilité, agitation extrême évoluant vers un délire et un coma).
Un traitement symptomatique devra être instauré.
La paroxétine ne doit pas être utilisée en association avec les précurseurs de la sérotonine (comme le L-tryptophane, l’oxitriptan) en raison du risque de syndrome sérotoninergique (voir rubriques 4.3 et 4.5).
Manie
Comme pour tous les antidépresseurs, la paroxétine doit être utilisée avec précaution chez les patients ayant des antécédents d’épisode maniaque. En cas de virage maniaque, le traitement par la paroxétine devra être arrêté.
Insuffisance rénale/hépatique
Une attention particulière est recommandée chez les patients présentant une insuffisance rénale sévère ou une insuffisance hépatique (voir rubrique 4.2).
Diabète
Les traitements par Inhibiteurs Sélectifs de la Recapture de la Sérotonine (ISRS) peuvent déséquilibrer le contrôle glycémique des patients diabétiques. L’adaptation des doses d’insuline et/ou de l’hypoglycémiant oral peut s’avérer nécessaire. De plus, des études suggèrent qu’une augmentation de la glycémie peut survenir lors de l’admnistration concomitante de paroxétine et pravastatine (voir rubrique 4.5).
Epilepsie
Comme d’autres antidépresseurs, la paroxétine doit être utilisée avec précaution chez les patients épileptiques.
Convulsions
L’incidence globale des crises convulsives est inférieure à 0,1 % chez les patients traités par la paroxétine. La survenue de crises convulsives impose l’arrêt du traitement.
Electroconvulsivothérapie (ECT)
Il existe peu de données cliniques sur l’administration concomitante de paroxétine et d’électroconvulsivothérapie.
Glaucome
Comme d’autres ISRS, la paroxétine peut provoquer une mydriase et devra être utilisée avec prudence chez les patients ayant un glaucome à angle étroit ou un antécédent de glaucome.
Pathologies cardiaques
Les précautions d’usage doivent être observées chez les patients présentant des pathologies cardiaques.
Allongement de l’intervalle QT
Des cas d’allongement de l’intervalle QT ont été rapportés pendant la période post-commercialisation.
La paroxétine doit être utilisée avec précaution chez les patients ayant des antécédents (familiaux) d’allongement de l’intervalle QT, les patients utilisant de façon concomitante des médicaments antiarythmiques ou d’autres médicaments pouvant potentiellement allonger l’intervalle QT, ou chez ceux ayant une maladie cardiaque préexistante telle qu’une insuffisance cardiaque, une cardiopathie ischémique, un bloc cardiaque ou des arythmies ventriculaires, une bradycardie, et une hypokaliémie ou une hypomagnésémie (voir rubriques 4.3 et 4.5).
Hyponatrémie
Une hyponatrémie a été rarement rapportée, principalement chez le sujet âgé.
Une attention particulière devra également être portée aux patients présentant un risque d’hyponatrémie lié à un traitement concomitant ou une cirrhose.
L’hyponatrémie est généralement réversible à l’arrêt de la paroxétine.
Hémorragies
Des saignements cutanés tels des ecchymoses et des purpuras ont été rapportés avec les ISRS. D’autres manifestations hémorragiques, telles des hémorragies gastro-intestinales et gynécologiques, ont été rapportées.
Le risque de saignement d’origine non menstruelle peut être accru chez les patients âgés.
Les ISRS/INSRS peuvent augmenter le risque d'hémorragie du post-partum (voir rubriques 4.6 et 4.8).La prudence est conseillée chez les patients traités simultanément par des ISRS et des anticoagulants oraux, des médicaments agissant sur la fonction plaquettaire ou d'autres médicaments susceptibles d'augmenter le risque de saignement (ex : antipsychotiques atypiques tels que la clozapine, les phénothiazines, la plupart des antidépresseurs tricycliques, l'aspirine, les AINS et les inhibiteurs de la COX-2) ainsi que chez les patients ayant des antécédents d'anomalies de l'hémostase ou souffrant de pathologies qui les prédisposent à des saignements (voir rubrique 4.8).
Interaction avec le tamoxifène
La paroxétine, puissant inhibiteur du CYP2D6, peut entrainer une diminution des concentrations d’endoxifène, l’un des plus importants métabolites actifs du tamoxifène. De ce fait, la paroxétine doit être évitée autant que possible pendant un traitement par tamoxifène (voir rubrique 4.5).
Symptômes de sevrage à l’arrêt du traitement par paroxétine
Les symptômes de sevrage à l’arrêt du traitement sont fréquents, particulièrement si l’arrêt est brutal (voir rubrique 4.8).
Dans les essais cliniques, des effets indésirables ont été observés lors de l’arrêt du traitement chez 30 % des patients traités par la paroxétine contre 20 % des patients recevant un placebo.
La survenue de symptômes de sevrage n’est pas synonyme d’addiction ou de dépendance.
Le risque de symptômes de sevrage peut être fonction de plusieurs facteurs incluant la durée du traitement, la posologie et le taux de réduction de la dose.
Ont été rapportés : sensations vertigineuses, troubles sensoriels (incluant paresthésies, et sensations à type de décharge électrique et acouphènes), troubles du sommeil (incluant rêves intenses), agitation ou anxiété, nausées, tremblement, confusion, hypersudation, céphalées, diarrhée, palpitations, instabilité émotionnelle, irritabilité et troubles visuels. Généralement, ces symptômes sont d’intensité légère à modérée, mais ils peuvent être d’intensité plus sévère chez certains patients.
Ils surviennent généralement dans les premiers jours suivant l’arrêt du traitement, mais quelques très rares cas ont été rapportés chez des patients ayant accidentellement sauté une prise. Généralement, ces symptômes sont spontanément résolutifs en deux semaines même si, chez certaines personnes, ils peuvent se prolonger (deux-trois mois, voire plus). Il est donc conseillé de diminuer progressivement la dose de paroxétine sur une période de plusieurs semaines ou mois, selon les besoins des patients (voir rubrique 4.2).
Attention, ce flacon contient une capsule déshydratante, ne pas l'avaler.
Dysfonction sexuelle
Les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS) pourraient causer des symptômes de dysfonction sexuelle (voir rubrique 4.8). Des cas de dysfonction sexuelle dont les symptômes se sont prolongés malgré l’arrêt du traitement par des ISRS ont été rapportés.
Excipient
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Comme avec les autres ISRS, l’association de la paroxétine à des médicaments sérotoninergiques peut entraîner une majoration des effets de la sérotonine (syndrome sérotoninergique : voir rubrique 4.4).
Une attention particulière et une surveillance clinique étroite sont recommandées lorsque ces médicaments sérotoninergiques (incluant L-tryptophane, triptans, tramadol), linézolide, chlorure de méthylthioninium (bleu de méthylène), ISRS, lithium, péthidine, buprénorphine et préparations à base de millepertuis – Hypericum perforatum) sont associés à la paroxétine. La prudence est également de mise avec le fentanyl, utilisé en anesthésie générale ou comme traitement de la douleur chronique.
L’utilisation en association de paroxétine et d’IMAOs est contre-indiquée en raison du risque de syndrome sérotoninergique (voir rubrique 4.3).
Pimozide
Une augmentation des concentrations de pimozide d’environ 2,5 fois en moyenne a été mise en évidence dans une étude d’interaction entre une faible dose de pimozide (2 mg) et une dose de 60 mg de paroxétine. Cela peut être dû aux propriétés inhibitrices connues de la paroxétine sur le CYP2D6. Le pimozide ayant un index thérapeutique étroit et pouvant entraîner un allongement de l’intervalle QT, l’association de paroxétine et de pimozide est contre-indiquée (voir rubrique 4.3).
Médicaments qui allongent l’intervalle QT
Le risque d'allongement de l'intervalle QTc et/ou d'arythmies ventriculaires (par exemple des torsades de pointe) peut être augmenté par l'utilisation concomitante d'autres médicaments qui allongent l'intervalle QTc (par exemple certains antipsychotiques) (voir rubrique 4.4). L'utilisation concomitante de la thioridazine et de la paroxétine est contre-indiquée car, comme pour d'autres médicaments qui inhibent l'enzyme hépatique CYP450 2D6, la paroxétine peut augmenter les taux plasmatiques de thioridazine, ce qui peut allonger l'intervalle QT (voir rubrique 4.3).
Enzymes du métabolisme
Le métabolisme et la pharmacocinétique de la paroxétine peuvent être modifiés par l’inhibition ou l’induction des enzymes la métabolisant.
Lorsque la paroxétine doit être associée à un inhibiteur enzymatique connu, les doses recommandées les plus faibles seront utilisées.
Aucun ajustement de dose n’est nécessaire lorsque la paroxétine est co-administrée avec des inducteurs enzymatiques (ex : carbamazépine, rifampicine, phénobarbital, phénytoïne) ou avec une association fosamprénavir/ritonavir. Tout ajustement de posologie de la paroxétine (soit après l’instauration du traitement, soit au décours de l’arrêt d’un inducteur enzymatique) sera basé sur l’effet clinique observé (tolérance et efficacité).
Curares
Les ISRS peuvent diminuer l’activité de la cholinestérase plasmatique induisant une prolongation de l’inhibition neuromusculaire du mivacurium et du suxamethonium.
Association fosamprénavir/ritonavir
La co-administration d’une association fosamprénavir/ritonavir à la posologie de 700/100 mg 2 fois par jour, avec 20 mg par jour de paroxétine, chez des volontaires sains pendant 10 jours, a entraîné une diminution significative de la concentration plasmatique de paroxétine d’environ 55 %. Lors de cette co-administration avec la paroxétine, les concentrations plasmatiques de fosamprénavir/ritonavir étaient similaires aux valeurs de référence issues d’autres études, indiquant que la paroxétine n’avait pas d’effet significatif sur le métabolisme de l’association fosamprénavir/ritonavir. Il n’y a pas de données disponibles sur les effets à long terme de la co-administration de paroxétine et d’une association fosamprénavir/ritonavir au-delà de 10 jours.
Procyclidine
L’administration journalière de paroxétine accroît significativement les concentrations plasmatiques de procyclidine. Si des effets anti-cholinergiques sont observés, la dose de procyclidine devra être réduite.
Anti-convulsivants
Carbamazépine, phénytoïne, valproate de sodium. L’administration concomitante ne semble pas avoir d’influence sur le profil pharmacocinétique/dynamique chez les patients épileptiques.
Inhibition du CYP2D6 par la paroxétine
Comme d’autres antidépresseurs, parmi lesquels d’autres ISRS, la paroxétine inhibe l’isoenzyme CYP2D6 du cytochrome P450 hépatique. L’inhibition de cette isoenzyme peut entraîner l’augmentation des concentrations plasmatiques des médicaments associés métabolisés par elle.
Ces médicaments comprennent certains antidépresseurs tricycliques (clomipramine, nortriptyline et désipramine), les neuroleptiques de type phénothiazine (ex : perphénazine et thioridazine, voir rubrique 4.3 et le paragraphe « Médicaments qui allongent l’intervalle QT » dans la rubrique 4.5 ci-dessus), la rispéridone, l’atomoxétine, certains antiarythmiques de type 1c (ex : propafénone et flécaïnide) et le métoprolol. Il n’est pas recommandé d’utiliser la paroxétine en association avec le métoprolol lorsqu’il est administré dans l’insuffisance cardiaque, en raison d’un index thérapeutique étroit du métoprolol dans cette indication.
Une interaction pharmacocinétique entre les inhibiteurs du CYP2D6 et le tamoxifène, montrant une diminution de 65-75% des concentrations plasmatiques d’endoxifène, une des formes les plus actives du tamoxifène, a été rapportée dans la littérature. Une diminution de l’efficacité du tamoxifène a été rapportée dans quelques études en cas d’utilisation concomitante de certains antidépresseurs ISRS. Une diminution de l’effet du tamoxifène ne pouvant être exclue, l’association d’un inhibiteur puissant du CYP2D6 (incluant la paroxétine) au tamoxifène doit être, autant que possible, évitée (voir rubrique 4.4).
Alcool
Comme avec les autres traitements psychotropes, les boissons alcoolisées sont déconseillées pendant le traitement.
Anticoagulants oraux
Une interaction pharmacodynamique peut se produire entre la paroxétine et les anticoagulants oraux. L’administration concomitante de paroxétine avec ces médicaments peut entraîner une augmentation de l’activité anticoagulante et du risque hémorragique. La paroxétine doit donc être utilisée avec prudence chez les patients traités par anticoagulants oraux (voir rubrique 4.4).
Anti-inflammatoires non stéroïdiens et acide acétylsalicylique, autres agents antiplaquettaires
Une interaction pharmacodynamique peut se produire entre la paroxétine et les AINS/acide acétylsalicylique. L’administration concomitante de ces médicaments peut augmenter le risque hémorragique (voir rubrique 4.4).
La prudence est conseillée chez les patients traités par des ISRS en association avec des anticoagulants oraux, des médicaments agissant sur la fonction plaquettaire ou susceptibles d'augmenter le risque de saignement (ex : antipsychotiques atypiques tels que la clozapine, les phénothiazines, la plupart des antidépresseurs tricycliques, l'aspirine, les AINS et les inhibiteurs de la COX-2) ainsi que chez les patients ayant des antécédents d'anomalies de l'hémostase ou souffrant de pathologies qui les prédisposent aux saignements.
Pravastatine
Une interaction entre la paroxétine et la pravastatine a été observée dans des études suggérant que l’administration concomittante de paroxétine et de pravastatine pouvait conduire à une augmentation de la glycémie. Un ajustement de la posologie des hypoglycémiants oraux et/ou de l’insuline peut s’avérer nécessaire chez les patients diabétiques recevant de la paroxétine et de la pravastatine (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
La paroxétine ne sera utilisée pendant la grossesse que si elle est strictement nécessaire. Le médecin devra évaluer l’intérêt d’un traitement alternatif chez une femme enceinte ou envisageant de l’être. Une interruption brutale du traitement doit être évitée au cours de la grossesse (voir rubrique 4.2).
Les données observationnelles ont montré un risque accru (moins de 2 fois) d'hémorragie du post-partum suite à une exposition aux ISRS/INSRS dans le mois précédant la naissance (voir rubriques 4.4 et 4.8).
Une surveillance du nouveau-né devra être effectuée si l’utilisation de la paroxétine est poursuivie jusqu’en fin de grossesse, particulièrement au troisième trimestre.
Les symptômes suivants peuvent survenir chez le nouveau-né après administration de paroxétine chez la mère pendant le troisième trimestre de la grossesse : détresse respiratoire, cyanose, apnée, convulsions, instabilité de la température, difficulté d’alimentation, vomissements, hypoglycémie, hypotonie, hypertonie, hyperréflexie, tremblements, nervosité, irritabilité, léthargie, pleurs permanents, somnolence et troubles du sommeil. Ces symptômes peuvent être dus soit à des effets sérotoninergiques soit à des symptômes de sevrage. Dans la majorité des cas, ces symptômes surviennent immédiatement ou presque après l’accouchement (moins de 24 heures).
Des données épidémiologiques semblent indiquer que l’utilisation d’ISRS pendant la grossesse, particulièrement en fin de grossesse, pourrait augmenter le risque d’hypertension artérielle pulmonaire persistante (HTAPP) chez le nouveau-né. Le risque observé était d’environ 5 cas pour 1000 grossesses. Dans la population générale, le risque d’HTAPP chez le nouveau-né est de 1 à 2 cas pour 1000 grossesses.
Les études chez l’animal ont montré une toxicité sur la reproduction, mais n’indiquent pas d’effets délétères directs sur la grossesse, le développement embryo/fœtal, l’accouchement ou le développement postnatal (voir rubrique 5.3).
Allaitement
De faibles quantités de paroxétine sont excrétées dans le lait maternel.
Dans les études publiées, les concentrations sériques des nourrissons allaités étaient indétectables (< 2 nanogrammes/ml) ou très faibles (< 4 nanogrammes/ml). Aucun signe d’un effet du médicament n’a été observé chez ces nourrissons.
Aucun effet n’étant attendu, l’allaitement est envisageable.
Fertilité
Des données chez l’animal ont montré que la paroxétine pourrait affecter la qualité du sperme (voir rubrique 5.3). Des données in vitro obtenues avec du matériel humain laissent suggérer une modification de la qualité du sperme ; cependant, des cas rapportés chez l’homme sous traitement par certains ISRSs (dont la paroxétine) ont montré que cet effet relatif à la qualité du sperme semble être réversible. L’impact sur la fertilité humaine n’a pas été observé jusqu’à présent.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Bien que la paroxétine n'augmente pas les atteintes mentales et motrices causées par l'alcool, l'utilisation concomitante de la paroxétine et de l'alcool est déconseillée.
Les effets indésirables sont listés ci-dessous par système organe et fréquence.
Les fréquences sont définies comme suit : très fréquents (≥ 1/10), fréquents (≥ 1/100, < 1/10), peu fréquents (≥ 1/1 000, < 1/100), rares (≥ 1/10 000, < 1/1 000) et très rares (< 1/10 000), indéterminés (ne pouvant être estimés avec les données disponibles).
Troubles hématologiques et du système lymphatique
Peu fréquents : saignements anormaux, principalement cutanéo-muqueux (incluant des ecchymoses et des saignements d’origine gynécologique), leucopénie.
Très rare : thrombocytopénie.
Troubles du système immunitaire
Très rares : réactions allergiques sévères et potentiellement fatales (incluant réactions anaphylactoïdes et œdème de Quincke).
Troubles endocriniens
Très rare : syndrome de sécrétion inappropriée de l’hormone anti-diurétique (SIADH).
Troubles du métabolisme et de la nutrition
Fréquents : augmentation de la cholestérolémie, diminution de l’appétit.
Peu fréquent : une modification du contrôle glycémique a été rapportée chez les patients diabétiques (voir rubrique 4.4).
Rare : hyponatrémie. La plupart des cas ont été décrits chez des patients âgés et sont parfois dus à un syndrome de sécrétion inappropriée de l’hormone anti-diurétique (SIADH).
Troubles psychiatriques
Fréquents : somnolence, insomnie, agitation, rêves anormaux (y compris cauchemars).
Peu fréquents : confusion, hallucinations.
Rares : réactions maniaques, anxiété, dépersonnalisation, attaques de panique, akathisie (voir rubrique 4.4).
Fréquence indéterminée : idées, comportements suicidaires, agression, bruxisme.
Des cas d’idées et de comportements suicidaires ont été rapportés durant le traitement par la paroxétine ou peu après son arrêt (voir rubrique 4.4).
*Des cas d’agression ont été observés lors de l’expérience après la mise sur le marché.
Ces symptômes peuvent également être dus à la pathologie sous-jacente.
Troubles du système nerveux
Fréquents : sensations vertigineuses, tremblements, céphalées, difficultés de concentration.
Peu fréquents : syndromes extrapyramidaux.
Rares : convulsions, syndrome des jambes sans repos.
Très rare : syndrome sérotoninergique (les symptômes peuvent inclure agitation, confusion, hypersudation, hallucinations, hyperréflexie, myoclonie, frissons, tachycardie et tremblements).
Des syndromes extra-pyramidaux incluant des dyskinésies bucco-faciales ont été rapportés chez des patients ayant parfois des mouvements anormaux sous-jacents ou chez des patients traités par des neuroleptiques.
Troubles oculaires
Fréquent : vision trouble.
Peu fréquent : mydriase (voir rubrique 4.4).
Très rare : glaucome aigu.
Troubles de l’oreille et du labyrinthe
Fréquence indéterminée : acouphènes.
Troubles cardiaques
Peu fréquent : tachycardie sinusale.
Rare : bradycardie.
Troubles vasculaires
Peu fréquents : élévations ou diminutions transitoires de la pression artérielle, hypotension orthostatique.
Des cas d’élévations ou de diminutions transitoires de la pression artérielle ont été rapportés à la suite d’un traitement par la paroxétine, habituellement chez des patients ayant une hypertension artérielle ou une anxiété pré-existantes.
Troubles respiratoires, thoraciques et médiastinaux
Fréquent : bâillements.
Troubles gastro-intestinaux
Très fréquents : nausées.
Fréquents : constipation, diarrhée, vomissements, sécheresse buccale.
Très rares : saignements gastro-intestinaux.
Fréquence indéterminée : colite microscopique
Troubles hépato-biliaires
Rare : élévation des enzymes hépatiques.
Très rares : atteintes hépatiques (telles que hépatites, parfois associées à un ictère et/ou une insuffisance hépatocellulaire).
Des cas d’élévation d’enzymes hépatiques ont été rapportés. Très rarement, des cas d’hépatites, parfois associées à un ictère et/ou une insuffisance hépatocellulaire ont été rapportés après la commercialisation de la paroxétine. En cas d’élévation prolongée des résultats des tests de la fonction hépatique, l’arrêt du traitement doit être envisagé.
Troubles cutanés et du tissu sous-cutané
Fréquent : hypersudation.
Peu fréquents : éruption cutanée, prurit.
Très rares : réactions cutanées graves (y compris érythème polymorphe, syndrome de Stevens-Johnson et nécrolyse épidermique toxique ou syndrome de Lyell), urticaire, réactions de photosensibilisation.
Troubles du rein et des voies urinaires
Peu fréquents : rétention urinaire, incontinence urinaire.
Troubles des organes de reproduction et du sein
Très fréquent : dysfonction sexuelle.
Rares : hyperprolactinémie/galactorrhée, troubles menstruels (incluant ménorragie, métrorragie, aménorrhée, retard de règles et règles irrégulières).
Très rare : priapisme.
Fréquence indéterminée : hémorragie du post-partum*.
Des hémorragies du post-partum ont été signalées pour la classe thérapeutique des ISRS/INSRS (voir rubriques 4.4 et 4.6).
Troubles musculo-squelettiques et systémiques
Rares : arthralgie, myalgie.
Des études épidémiologiques, menées principalement chez des patients âgés de 50 ans et plus, indiquent une augmentation du risque de fractures chez les patients qui prennent des ISRS et des antidépresseurs tricycliques. Le mécanisme entrainant ce risque n’est pas connu.
Troubles généraux et anomalies au site d’administration
Fréquents : asthénie, prise de poids.
Très rares : œdèmes périphériques.
Symptômes de sevrage à l’arrêt du traitement
Fréquents : sensations vertigineuses, troubles sensoriels, troubles du sommeil, anxiété, céphalées.
Peu fréquents : agitation, nausées, tremblements, confusion, hypersudation, instabilité émotionnelle, troubles visuels, palpitations, diarrhée, irritabilité.
L’arrêt du traitement par la paroxétine, particulièrement quand il est brutal, entraîne fréquemment des symptômes de sevrage.
Ont été observés : sensations vertigineuses, troubles sensoriels (incluant paresthésies et sensations à type de décharges électriques et acouphènes), troubles du sommeil (incluant rêves intenses), agitation ou anxiété, nausées, tremblements, confusion, hypersudation, céphalées, diarrhée, palpitations, instabilité émotionnelle, irritabilité et troubles visuels.
Généralement ces effets sont d’intensité légère à modérée et spontanément résolutifs ; cependant, chez certains patients, ils peuvent être sévères et/ou prolongés.
Il est donc recommandé de diminuer progressivement les doses de paroxétine lorsque le traitement n’est plus nécessaire (voir rubriques 4.2 et 4.4).
EFFETS INDESIRABLES AU COURS DES ESSAIS CLINIQUES PEDIATRIQUES
Les évènements indésirables suivants ont été observés :
Une augmentation des comportements suicidaires (y compris des tentatives de suicides et des pensées suicidaires), des comportements auto-agressifs et une hostilité plus importante. Des pensées suicidaires et des tentatives de suicide ont été principalement observées dans le cadre d’études cliniques portant sur des adolescents présentant un trouble dépressif majeur. Une hostilité accrue survenait en particulier chez des enfants présentant des troubles obsessionnels compulsifs et principalement chez des enfants de moins de 12 ans.
Autres évènements observés : diminution de l’appétit, tremblements, transpiration excessive, hyperkinésie, agitation, labilité émotionnelle (y compris pleurs et humeur fluctuante), évènements indésirables en relation avec des saignements, principalement au niveau de la peau et des muqueuses.
Les évènements observés après l’arrêt / la diminution de la paroxétine sont les suivants : labilité émotionnelle (y compris pleurs, modifications de l’humeur, auto-agressivité, pensées suicidaires et tentatives de suicide), nervosité, sensations vertigineuses, nausées et douleurs abdominales (voir rubrique 4.4).
Voir rubrique 5.1 pour de plus amples informations sur les études cliniques pédiatriques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Les informations disponibles sur les cas de surdosage à la paroxétine démontrent qu'il existe une marge de sécurité importante.
Lors de surdosages avec la paroxétine, à côté des symptômes mentionnés en rubrique 4.8, les symptômes suivants ont été observés : fièvre et contractions musculaires involontaires. Les patients se sont généralement rétablis sans séquelles sérieuses, même dans les cas où des doses allant jusqu'à 2000 mg avaient été prises, seules.
Des effets tels que coma ou modifications de l'ECG, d'évolution très rarement fatale, ont été rapportés occasionnellement, généralement en cas de polyintoxications avec d'autres médicaments psychotropes, voire de l'alcool.
Traitement
Il n'existe pas d'antidote spécifique de la paroxétine.
Le traitement comporte les mêmes mesures générales que pour tout surdosage avec des antidépresseurs.
L'administration de 20 à 30 g de charbon activé peut être envisagé, si possible dans les heures qui suivent le surdosage, pour diminuer l'absorption de paroxétine.
Une surveillance régulière des constantes vitales et une observation étroite des patients sont indiquées. La prise en charge sera fonction de l'état clinique du patient.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
La paroxétine est un inhibiteur puissant et sélectif de la recapture de la 5-hydroxytryptamine (5HT, sérotonine). Son action antidépressive et son efficacité dans le traitement des Troubles Obsessionnels Compulsifs, du trouble Anxiété Sociale/Phobie Sociale, du trouble Anxiété Généralisée, de l’Etat de stress Post-Traumatique et du trouble Panique semblent être dues à son inhibition spécifique de la recapture de la sérotonine dans les neurones cérébraux.
La paroxétine n’est pas chimiquement apparentée aux antidépresseurs tricycliques, tétracycliques et autres antidépresseurs disponibles.
La paroxétine a une faible affinité pour les récepteurs muscariniques cholinergiques et les études effectuées sur l’animal n’ont montré qu’une faible activité anticholinergique.
En relation avec cette action sélective, des études in vitro ont montré que, contrairement à la plupart des antidépresseurs tricycliques, la paroxétine a peu d’affinité pour les récepteurs alpha 1, alpha 2, et bêta adrénergiques, dopaminergiques (D2), 5HT1 apparentés, 5-HT2 et histaminergiques (H1). Cette absence d’interaction avec les récepteurs post-synaptiques in vitro est corroborée par des études in vivo qui démontrent l’absence d’effet dépresseur sur le système nerveux central ainsi que de propriétés hypotensives.
Effets pharmacodynamiques
La paroxétine n’altère pas les fonctions psychomotrices et ne potentialise pas les effets dépresseurs de l’éthanol.
Comme avec les autres ISRS, la paroxétine entraîne des symptômes de stimulation excessive des récepteurs à la sérotonine lorsqu’elle est administrée chez l’animal ayant reçu au préalable des inhibiteurs de la monoamine oxydase (IMAO) ou du tryptophane.
Les études comportementales et électroencéphalographiques (EEG) montrent que la paroxétine est faiblement activatrice à des doses généralement supérieures à celles entraînant l’inhibition de la recapture de la sérotonine. Ces propriétés activatrices ne sont pas de nature amphétaminique.
Les études chez l’animal indiquent que la paroxétine est bien tolérée au niveau cardiovasculaire. Chez le volontaire sain, la paroxétine n’entraîne pas de modification cliniquement significative de la pression artérielle, de la fréquence cardiaque et de l’électrocardiogramme.
Contrairement aux antidépresseurs qui inhibent la recapture de la noradrénaline, les études indiquent que la paroxétine a une faible propension à inhiber les effets anti-hypertenseurs de la guanéthidine.
Dans le traitement des troubles dépressifs, la paroxétine montre une efficacité comparable aux antidépresseurs standards.
La paroxétine peut avoir un intérêt thérapeutique chez les patients ne répondant pas aux thérapeutiques standards.
La prise matinale de la paroxétine n’a pas d’effet préjudiciable sur la qualité ou la durée du sommeil.
De plus, les patients sont susceptibles d’améliorer leur sommeil quand ils répondent au traitement par la paroxétine.
Analyse de la suicidalité chez l’adulte
Une analyse spécifique des études comparant la paroxétine à un placebo chez des adultes présentant des troubles psychiatriques a montré une fréquence de comportement suicidaire plus élevée chez les jeunes adultes (âgés de 18 à 24 ans) traités par paroxétine que chez ceux recevant un placebo (2,19 % vs 0,92 %). Aucune différence de ce type n’a été observée dans les groupes de sujets plus âgés. Chez les adultes présentant un épisode dépressif majeur (tous âges confondus), une augmentation de la fréquence des comportements suicidaires a été observée chez les patients traités par paroxétine, par rapport à ceux recevant un placebo (0,32 % vs 0,05 %) ; tous les événements observés étaient des tentatives de suicide. Cependant, la majorité des tentatives observées sous paroxétine (8 sur 11) concernaient des adultes plus jeunes (voir rubrique 4.4).
Dose réponse
Dans les études à dose fixe, la courbe de dose-réponse est aplatie, suggérant l’absence de bénéfice à utiliser des doses supérieures aux doses recommandées en termes d’efficacité. Cependant, quelques données cliniques suggèrent que l’augmentation des doses pourrait être bénéfique chez certains patients.
Efficacité long terme
L’efficacité à long terme de la paroxétine dans la dépression a été démontrée dans une étude de maintien d’efficacité sur 52 semaines (suivant un schéma de type « prévention des rechutes ») : 12 % des patients recevant de la paroxétine (20-40 mg par jour) ont rechuté versus 28 % des patients dans le bras placebo.
L’efficacité à long terme de la paroxétine dans les troubles obsessionnels compulsifs a été démontrée par 3 études de maintien d’efficacité sur 24 semaines, de type « prévention des rechutes ». L’une des 3 études a montré une différence significative entre la proportion des rechutes sous paroxétine (38 %) et celle sous placebo (59 %).
L’efficacité à long terme de la paroxétine dans le traitement du trouble Panique a été démontrée par une étude de maintien d’efficacité sur 24 semaines de type « prévention des rechutes » : 5 % des patients sous paroxétine (10-40 mg) ont rechuté versus 30 % des patients sous placebo. Cela a été confirmé dans une étude de maintien d’efficacité sur 36 semaines.
L’efficacité à long terme de la paroxétine dans le traitement du trouble Anxiété Sociale, du trouble Anxiété Généralisée et de l’Etat de stress Post-Traumatique n’a pas été suffisamment démontrée.
Effets indésirables au cours des essais cliniques pédiatriques
Au cours d’essais cliniques à court terme (jusqu’à 10-12 semaines) chez l’enfant et l’adolescent, les effets indésirables suivants ont été observés chez les patients traités par la paroxétine, avec une fréquence ≥ 2 % et au moins deux fois supérieure à celle observée dans le groupe placebo : augmentation des comportements suicidaires (incluant tentatives de suicide et pensées suicidaires), comportements auto-agressifs et hostilité accrue. Pensées suicidaires et tentatives de suicide ont été principalement observées au cours des essais cliniques chez des adolescents atteints d’épisodes dépressifs majeurs. L’augmentation de l’hostilité a notamment été observée chez les enfants présentant des troubles obsessionnels compulsifs en particulier chez les enfants de moins de 12 ans. Les autres effets indésirables observés plus souvent dans le groupe paroxétine comparativement au groupe placebo étaient : diminution de l’appétit, tremblement, hypersudation, hyperkinésie, agitation, labilité émotionnelle (incluant pleurs et fluctuations de l’humeur).
Dans les études comportant un schéma d’arrêt progressif du traitement, les symptômes rapportés durant la phase de réduction de posologie ou à l’arrêt du traitement, avec une fréquence ≥ à 2 % et au moins double de celle observée dans le groupe placebo étaient : labilité émotionnelle (incluant pleurs, fluctuations de l’humeur, auto-agressivité, pensées suicidaires et tentatives de suicide), nervosité, sensations vertigineuses, nausées et douleurs abdominales (voir rubrique 4.4).
Dans cinq études en groupes parallèles comportant une durée de traitement allant de 8 semaines à 8 mois, des évènements indésirables en relation avec des saignements touchant principalement la peau et les muqueuses, ont été observés chez des patients traités par la paroxétine à une fréquence de 1,74 %, tandis que leur fréquence dans le groupe traité par placebo était de 0,74 %.
5.2. Propriétés pharmacocinétiques
La paroxétine est bien absorbée par voie orale et subit un effet de premier passage hépatique. En raison de cet effet, la quantité de paroxétine présente dans la circulation systémique est inférieure à celle absorbée par le tractus gastro-intestinal. Une saturation partielle de l'effet de premier passage hépatique et une diminution de la clairance plasmatique surviennent quand l'exposition de l'organisme au produit augmente après la prise de doses uniques plus élevées ou de doses répétées. Il en résulte une augmentation disproportionnée des concentrations plasmatiques, entraînant des paramètres pharmacocinétiques non constants et par conséquent, une cinétique non linéaire du produit. Cependant, cette non linéarité est généralement faible et limitée aux sujets présentant des taux plasmatiques bas lors de l'administration de faibles doses.
Les concentrations plasmatiques à l'équilibre sont atteintes après 7 à 14 jours de traitement avec les formes à libération immédiate ou prolongée et les paramètres restent stables lors d'un traitement à long terme.
Distribution
La paroxétine est très largement distribuée dans les tissus et les résultats de pharmacocinétique montrent que seulement 1 % de la paroxétine absorbée reste dans le compartiment plasmatique.
Environ 95 % de la paroxétine présente est fixée aux protéines plasmatiques aux concentrations thérapeutiques.
Aucune corrélation n'a été démontrée entre les concentrations plasmatiques de paroxétine et les effets cliniques observés (effets indésirables et efficacité).
Biotransformation
Les principaux métabolites de la paroxétine sont des produits polaires et conjugués d'oxydation et de méthylation, facilement éliminés. Considérant leur faible activité pharmacologique, il est peu probable qu'ils contribuent aux effets thérapeutiques de la paroxétine.
Le métabolisme de la paroxétine ne compromet pas l'action sélective de la paroxétine sur la recapture de la sérotonine.
Élimination
L'élimination urinaire de la paroxétine sous forme inchangée représente généralement moins de 2 % de la dose initiale tandis que celle des métabolites atteint environ 64 %. Environ 36 % de la dose, dont moins de 1 % est sous forme inchangée, est éliminée dans les fèces, probablement par voie biliaire.
L'élimination de la paroxétine s'effectue donc presque entièrement sous forme métabolisée.
L'élimination des métabolites est biphasique : elle résulte initialement du premier passage hépatique, puis d'une élimination systémique de la paroxétine.
La demi-vie d'élimination est variable, mais généralement de 24 heures.
Populations particulières
Population âgée et Insuffisants rénaux/ hépatiques
Chez les sujets âgés, les insuffisants rénaux sévères et les insuffisants hépatiques, on observe une augmentation des concentrations plasmatiques de paroxétine, qui demeurent cependant dans les limites de celles observées chez les sujets adultes sains.
5.3. Données de sécurité préclinique
Carcinogénèse : la paroxétine n’a pas montré d’effet carcinogène lors d’études réalisées sur 2 ans chez le rat et la souris.
Génotoxicité : aucun effet génotoxique n’a été observé au cours de tests réalisés in vitro et in vivo.
Des études de toxicité sur la reproduction chez le rat ont montré que la paroxétine affecte la fertilité chez le mâle et la femelle en réduisant l’indice de fertilité et le taux de grossesse. Chez les rats, une augmentation de la mortalité des jeunes ainsi qu’un retard d’ossification ont été observés. Ces derniers effets sont probablement liés à la toxicité maternelle et ne sont pas considérés comme des effets directs sur le fœtus et/ou le nouveau-né.
Pelliculage : talc, dioxyde de titane (E171), copolymère de méthacrylate de butyle, de (2-diméthylaminoéthyl) méthacrylate et de méthylméthacrylate 1: 2: 1 (EUDRAGIT E 100).
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
6.5. Nature et contenu de l'emballage extérieur
14, 28, 30, 50, 100 ou 200 comprimés pelliculés sécables en flacon (PE) avec bouchon (PP).
14, 15, 28, 30, 50 ou 90 comprimés pelliculés sécables sous plaquette (Aluminium/Aluminium).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d'exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1 RUE DE TURIN
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 359 987 1 2 : 14 comprimés en flacon (PE)
· 34009 359 988 8 0 : 28 comprimés en flacon (PE)
· 34009 359 989 4 1 : 30 comprimés en flacon (PE)
· 34009 359 990 2 3 : 50 comprimés en flacon (PE)
· 34009 359 991 9 1 : 100 comprimés en flacon (PE)
· 34009 359 992 5 2 : 200 comprimés en flacon (PE)
· 34009 374 173 1 0 : 14 comprimés sous plaquette (Aluminium/Aluminium)
· 34009 374 174 8 8 : 15 comprimés sous plaquette (Aluminium/Aluminium)
· 34009 374 175 4 9 : 28 comprimés sous plaquette (Aluminium/Aluminium)
· 34009 374 176 0 0 : 30 comprimés sous plaquette (Aluminium/Aluminium)
· 34009 374 177 7 8 : 50 comprimés sous plaquette (Aluminium/Aluminium)
· 34009 374 178 3 9 : 90 comprimés sous plaquette (Aluminium/Aluminium)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 24/07/2025
PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable
Paroxétine (sous forme de chlorhydrate de paroxétine anhydre)
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ?
3. Comment prendre PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ET DANS QUELS CAS EST-IL UTILISE ?
PAROXETINE VIATRIS est un traitement destiné aux adultes souffrant de dépression et/ou de troubles anxieux.
Les troubles anxieux dans lesquels PAROXETINE VIATRIS peut être prescrit sont les suivants :
· troubles obsessionnels compulsifs (pensées répétitives, obsessionnelles avec comportement incontrôlable),
· trouble panique (attaques de panique, y compris celles causées par la peur des lieux publics, l'agoraphobie)
· trouble anxiété sociale (peur ou rejet de situations où vous devez être en société)
· état de stress post-traumatique (anxiété causée par un événement traumatique)
· anxiété généralisée.
PAROXETINE VIATRIS appartient à la classe de médicaments appelés Inhibiteurs Sélectifs de la Recapture de la Sérotonine (ISRS).
Le mécanisme d'action de PAROXETINE VIATRIS et des autres ISRS n'est pas complètement connu, mais ils augmenteraient le taux de sérotonine dans le cerveau.
Bien traiter votre dépression ou votre trouble anxieux est important pour vous aider à vous sentir mieux.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ?
Ne prenez jamais PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable :
· Si vous prenez ou avez pris au cours des 2 dernières semaines un médicament appelé IMAO (inhibiteur de la monoamine oxydase, incluant le moclobémide et le chlorure de méthylthioninium (bleu de méthylène)). Votre médecin vous dira comment débuter le traitement avec la paroxétine une fois que vous aurez arrêté le traitement par IMAO.
· Si vous prenez un anti-psychotique appelé thioridazine ou un anti-psychotique appelé pimozide.
· Si vous êtes allergique à la paroxétine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· Si vous êtes concerné par l’un de ces points, ne prenez pas PAROXETINE VIATRIS et informez-en votre médecin.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant de prendre PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable :
· Si vous prenez un autre traitement (voir rubrique ci-dessous « Autres médicaments et PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable »).
· Si vous prenez du tamoxifène pour traiter un cancer du sein. PAROXETINE VIATRIS pouvant diminuer l’efficacité du tamoxifène, votre médecin pourra préférer un autre traitement antidépresseur.
· Si vous avez des problèmes de reins, de foie ou de cœur.
· Si vous avez une anomalie du tracé cardiaque sur un électrocardiogramme (ECG) connue sous le nom d'intervalle QT prolongé.
· Si vous avez des antécédents familiaux d’allongement de l’intervalle QT, de maladies cardiaques telles qu’une insuffisance cardiaque, une fréquence cardiaque basse ou de faibles taux de potassium ou de magnésium.
· Si vous souffrez d'épilepsie ou si vous avez eu dans le passé des convulsions ou des crises.
· Si vous avez déjà eu un épisode « maniaque » (excitation incontrôlable et hyperactivité).
· Si vous avez eu une sismothérapie (électrochoc).
· Si vous avez des antécédents de troubles de la coagulation, des ecchymoses (bleus) ou si vous saignez facilement, ou si vous prenez un médicament qui peut augmenter les saignements comme la warfarine, des antipsychotiques comme la perphénazine ou la clozapine, des antidépresseurs tricycliques, des médicaments contre la douleur ou l'inflammation appelés Anti-inflammatoires Non Stéroïdiens (AINS) comme l'aspirine, l'ibuprofène, le célécoxib, l'étodolac, le diclofénac, le méloxicam.
· Si vous êtes diabétique.
· Si vous suivez un régime pauvre en sel.
· Si vous souffrez de glaucome (hypertension au niveau de l'œil).
· Si vous êtes enceinte ou envisagez de l'être ou si vous allaitez (voir rubriques « Grossesse, allaitement et fertilité »).
· Si vous avez moins de 18 ans (voir rubrique ci-dessous « Enfants et adolescents »).
Si vous présentez l’une des conditions ci-dessus et si vous n’en avez pas déjà parlé à votre médecin, retournez voir votre médecin et demandez-lui son avis sur la prise de la PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable.
Enfants et adolescents
PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ne doit habituellement pas être utilisé chez les enfants et les adolescents de moins de 18 ans.
Les patients de moins de 18 ans présentent un risque accru d’effets indésirables tels que tentative de suicide, pensées suicidaires ou comportement hostile (comportement agressif, d’opposition et colère) lorsqu’ils sont traités par PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable.
Néanmoins, il est possible que votre médecin décide de prescrire PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable à des patients de moins de 18 ans s’il/elle décide que c’est dans l’intérêt du patient.
Si votre médecin vous a prescrit (ou à votre enfant) PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable et que vous désirez en discuter avec lui, adressez-vous à lui. Vous devez informer votre médecin si l’un des symptômes ci-dessus apparaît ou s’aggrave lors de la prise de PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable par un patient de moins de 18 ans.
Dans cette tranche d’âge, la sécurité à long terme de PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable concernant la croissance, la maturation et le développement cognitif et comportemental n’a pas encore été établie.
Dans les études chez des patients de moins de 18 ans, les effets secondaires fréquents qui affectaient moins de 1 enfant/adolescent sur 10 étaient les suivants : augmentation des pensées suicidaires et des tentatives de suicide, agressivité envers soi-même, comportement hostile, agressif ou inamical, manque d’appétit, tremblements, transpiration excessive, hyperactivité, agitation, des fluctuations de l’humeur et accès de pleurs et des ecchymoses ou des saignements inhabituels (par exemple des saignements de nez). Dans ces études, les symptômes décrits ci-dessus ont également affecté les enfants et adolescents qui avaient reçu du placebo, mais à une fréquence moindre.
Dans ces études, certains patients de moins de 18 ans ont présenté des effets indésirables lors de la diminution de doses ou de l’arrêt de PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable. Ces effets étaient similaires à ceux observés chez l’adulte à l’arrêt de PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable (voir rubrique 3).
De plus, les patients de moins de 18 ans ont présenté fréquemment (moins de 1 patient sur 10) des douleurs abdominales, une nervosité, et des fluctuations de l’humeur (accès de pleurs, agressivité envers soi-même, pensées suicidaires et tentatives de suicide).
Idées suicidaires et aggravation de votre dépression ou de votre trouble anxieux
Si vous souffrez de dépression et/ou de troubles anxieux, vous pouvez parfois avoir des idées d'auto-agression (agression envers vous-même) ou de suicide. Ces manifestations peuvent être majorées au début d'un traitement par antidépresseur, car ce type de médicament n'agit pas tout de suite mais seulement après 2 semaines ou plus de traitement.
Vous êtes plus susceptible de présenter ce type de manifestations dans les cas suivants :
· si vous avez déjà eu des idées suicidaires ou d'auto-agression dans le passé ;
· si vous êtes un jeune adulte. Les études cliniques ont montré que le risque de comportement suicidaire était accru, chez les adultes de moins de 25 ans présentant une maladie psychiatrique et traités par antidépresseur.
Si vous avez des idées suicidaires ou d'auto-agression, contactez immédiatement votre médecin ou allez directement à l'hôpital.
Vous pouvez vous faire aider par un ami ou un parent, en lui expliquant que vous êtes dépressif ou que vous souffrez d'un trouble anxieux, et en lui demandant de lire cette notice. Vous pouvez lui demander de vous signaler s'il pense que votre dépression ou votre anxiété s'aggrave, ou s'il s'inquiète d'un changement dans votre comportement.
Effets indésirables importants observés avec PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable
Des patients traités par PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable développent une réaction appelée akathisie, qui comprend le fait de se sentir agité et de ne pas pouvoir rester assis ou debout tranquillement. D’autres patients développent un syndrome sérotoninergique ou syndrome malin des neuroleptiques, comportant certains ou la totalité des symptômes suivants : sensation de grande agitation ou d’irritabilité, sensation de confusion, d’agitation, sensation de chaleur, transpiration, tremblement, frissons, hallucinations (vision ou sons étranges), rigidité des muscles, contractions involontaires des muscles ou rythme cardiaque accéléré. La sévérité de ces symptômes peut s’aggraver conduisant à une perte de connaissance. Si vous remarquez un de ces symptômes, contactez votre médecin. Pour plus d’information sur les effets indésirables de PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable, lire ci-dessous la rubrique 4.
Les médicaments comme PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable (appelés ISRS) pourraient causer des symptômes de dysfonction sexuelle (voir rubrique 4). Dans certains cas, ces symptômes se sont prolongés après l’arrêt du traitement.
Autres médicaments et PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Certains médicaments peuvent modifier l’action de PAROXETINE VIATRIS, et augmenter le risque d’effets secondaires. PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable peut également modifier l’action d’autres médicaments.
Ces médicaments incluent :
· des médicaments appelés inhibiteurs de la monoamine oxydase (IMAO, incluant le moclobémide et le chlorure de méthylthioninium (bleu de méthylène)) ;
· des médicaments connus pour augmenter le risque de modifications de l’activité électrique du cœur (par exemple les antipsychotiques tels que la thioridazine ou le pimozide)
· l’aspirine, l’ibuprofène et d’autres médicaments appelés AINS (Anti-Inflammatoires Non Stéroïdiens) comme le célécoxib, l’étodolac, le diclofénac et le méloxicam, utilisés contre la douleur et l’inflammation
· le tramadol, la buprénorphine et la péthidine, des médicaments contre la douleur
· la buprénorphine combinée à la naloxone, un médicament utilisé dans la dépendance aux opioïdes
· des médicaments appelés triptans, comme le sumatriptan, indiqués pour traiter la migraine ;
· d’autres antidépresseurs incluant des ISRS et des antidépresseurs tricycliques comme la clomipramine, la nortriptyline et la désipramine ;
· un complément alimentaire appelé tryptophane ;
· mivacurium et suxaméthonium (utilisés en anesthésie)
· des médicaments tels que le lithium, la rispéridone, la perphénazine, la clozapine (appelés anti-psychotiques) utilisés pour traiter certaines affections psychiatriques
· le fentanyl, utilisé en anesthésie ou pour traiter les douleurs chroniques
· l’association fosamprénavir et ritonavir, utilisée pour le traitement de l’infection à VIH
· le millepertuis, une plante médicinale pour le traitement de la dépression
· le phénobarbital, la phénytoïne, le valproate de sodium ou la carbamazépine utilisés pour traiter l’épilepsie
· l’atomoxétine, qui est utilisée dans les troubles de l’attention avec hyperactivité
· la procyclidine, qui est utilisée dans le traitement de la maladie de Parkinson ou d’autres tremblements
· la warfarine ou d’autres médicaments appelés anticoagulants, utilisés pour fluidifier le sang
· la propafénone, la flécaïnide, médicaments utilisés dans les troubles du rythme cardiaque (arythmie)
· le métoprolol, un bêta-bloquant pour traiter l’hypertension et d’autres problèmes cardiaques
· la pravastatine, utilisée pour traiter un taux de cholestérol élevé
· la rifampicine, utilisée pour traiter la tuberculose et la lèpre
· le linézolide, un antibiotique
· le tamoxifène, utilisé pour le traitement du cancer du sein.
PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable avec des aliments, boissons et de l’alcool
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Grossesse
Chez les nourrissons dont les mères avaient pris PAROXETINE VIATRIS durant les premiers mois de la grossesse, quelques études ont montré une augmentation du risque de malformations à la naissance, en particulier cardiaques. Dans la population générale, le risque d’anomalie cardiovasculaire à la naissance est de 1%. Ce risque augmente jusqu’à 2% chez les mères ayant pris PAROXETINE VIATRIS. Si vous êtes enceinte, votre médecin et vous-même déciderez s’il est préférable pour vous de changer de traitement et d’arrêter progressivement ou de continuer PAROXETINE VIATRIS.
Assurez-vous que votre médecin ou la sage-femme soit informé que vous prenez PAROXETINE VIATRIS.
Si vous prenez PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable vers la fin de votre grossesse, il peut y avoir un risque accru de saignements vaginaux abondants peu après la naissance, surtout si vous avez déjà eu des problèmes de saignements. Votre médecin ou votre sage-femme doit savoir que vous prenez PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable afin de pouvoir vous conseiller. Si PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable est utilisé pendant la grossesse, particulièrement à la fin, PAROXETINE VIATRIS peut augmenter le risque de survenue de maladie grave chez les nouveau-nés, appelée risque d’hypertension pulmonaire persistante chez le nouveau-né. Dans ce cas, la pression du sang dans les vaisseaux entre le cœur et les poumons du nouveau-né est trop élevée. Si vous prenez PAROXETINE VIATRIS durant les 3 derniers mois de grossesse, votre nouveau-né peut également présenter d’autres symptômes, habituellement dans les 24 heures suivant la naissance. Ces symptômes sont :
· difficulté respiratoire,
· coloration violacée de la peau, des lèvres ou des ongles, ou difficulté de régulation de la température,
· vomissements ou difficultés d’alimentation,
· somnolence ou grande fatigue, sommeil perturbé ou pleurs permanents,
· rigidité ou relâchement anormal des muscles,
· nervosité, irritabilité, tremblements, convulsions,
· réflexes exagérés.
Si votre bébé présente l’un de ces symptômes, ou si vous vous interrogez sur la santé de votre bébé, parlez-en à votre médecin ou à la sage-femme. Ils vous indiqueront ce qu’il faut faire.
Allaitement
PAROXETINE VIATRIS passe dans le lait maternel en très faible quantité.
Si vous prenez PAROXETINE VIATRIS demandez conseil à votre médecin ou à votre pharmacien avant d’allaiter. Vous et votre médecin pourrez décider d’allaiter pendant votre traitement par PAROXETINE VIATRIS.
Fertilité
Des études chez l’animal ont montré que la paroxétine réduisait la qualité du sperme. Théoriquement, la fertilité pourrait être affectée, cependant l’impact sur la fertilité humaine n’a pas été observé à ce jour.
Conduite de véhicules et utilisation de machines
Les effets indésirables possibles de PAROXETINE VIATRIS incluent vertiges, confusion, somnolence ou vision trouble.
Si vous présentez ces effets secondaires, ne conduisez pas ou n'utilisez pas de machine.
PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable contient du sodium.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ?
Veillez à toujours prendre ce médicament en suivant exactement les instructions de cette notice ou les indications votre médecin, pharmacien ou infirmier/ère. Vérifiez auprès de votre médecin, pharmacien ou infirmier/ère en cas de doute.
Posologie
Parfois vous aurez besoin de prendre plus d’un comprimé ou une moitié de comprimé.
Les doses usuelles selon les pathologies sont indiquées dans le tableau ci-dessous.
|
Dose d’instauration de traitement |
Dose quotidienne recommandée |
Dose maximale quotidienne |
|
|
Dépression |
20 mg |
20 mg |
50 mg |
|
Troubles obsessionnels compulsifs |
20 mg |
40 mg |
60 mg |
|
Troubles paniques |
10 mg |
40 mg |
60 mg |
|
Trouble anxiété sociale |
20 mg |
20 mg |
50 mg |
|
Stress post-traumatique |
20 mg |
20 mg |
50 mg |
|
Trouble anxiété généralisée |
20 mg |
20 mg |
50 mg |
Mode d’administration
Prenez votre médicament de préférence le matin au cours du petit déjeuner.
Avaler les comprimés avec un verre d’eau.
Votre médecin vous indiquera la dose à prendre lorsque vous commencerez votre traitement par PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable. La plupart des patients commencent à se sentir mieux au bout de 2 semaines. Si vous ne commencez pas à vous sentir mieux à ce moment, contactez votre médecin qui vous conseillera. Il pourra décider d’augmenter les doses progressivement, par palier de 10 mg, jusqu’à la dose maximale quotidienne.
Ne prenez jamais plus de comprimés que ce que votre médecin vous a prescrit.
Ne les mâchez pas.
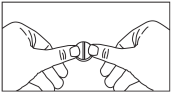
Les comprimés sont sécables, c’est-à-dire que vous pouvez, si besoin, les couper en 2 parts égales. Pour ceci, placer le comprimé sur une surface dure, placer un pouce ou index de chaque côté de la barre de sécabilité, appliquer une pression sur le côté du comprimé jusqu’à qu’il se divise en 2, selon le schéma ci-dessous :
Durée du traitement
Votre médecin vous indiquera la durée (plusieurs mois ou plus) pendant laquelle vous devrez prendre PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable.
Patients âgés
Si vous avez plus de 65 ans, la dose maximale recommandée est de 40 mg par jour.
Patients ayant une maladie des reins ou du foie
Si vous avez un problème rénal sévère ou de foie, votre médecin pourra vous prescrire une dose plus faible.
Si vous avez pris plus de PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable que vous n’auriez dû
Consultez immédiatement votre médecin ou votre pharmacien. Les effets secondaires possibles en cas de surdosage sont ceux listés en rubrique 4. « Quels sont les effets indésirables éventuels ? » ou les suivants : fièvre, contraction involontaire des muscles.
Si vous oubliez de prendre PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable
Prenez votre médicament au même moment tous les jours.
Si vous vous en rendez compte avant le coucher, prenez la dose de PAROXETINE VIATRIS.
Poursuivez le traitement comme d’habitude le lendemain.
Si vous vous en rendez compte pendant la nuit, ou le jour suivant, laissez de côté la dose oubliée et poursuivez le traitement comme d’habitude. Vous pouvez éventuellement avoir des symptômes de sevrage, mais ceux-ci disparaîtront lorsque vous prendrez la prochaine dose à l’heure habituelle.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Que faire si vous ne vous sentez pas mieux
PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ne soulagera pas vos symptômes immédiatement. Tous les antidépresseurs mettent du temps pour agir. Après 2 semaines, la plupart des personnes commencent à se sentir mieux, mais pour d’autres cela peut être plus long. Certaines personnes prenant des antidépresseurs se sentent plus mal avant de voir leur état s’améliorer. Si vous ne vous sentez pas mieux au bout de 2 semaines, consultez votre médecin qui vous conseillera et qui pourra éventuellement augmenter les doses. Votre médecin pourra vous demander de vous revoir au bout de 2 semaines après l’instauration de traitement. Informez votre médecin si vous ne commencez pas à vous sentir mieux.
Si vous arrêtez de prendre PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable
N'ARRETEZ pas le traitement avec PAROXETINE VIATRIS tant que le médecin ne vous l'a pas indiqué. Votre médecin vous conseillera, dans la plupart des cas, de réduire progressivement la dose sur plusieurs semaines.
Lorsque vous arrêtez de prendre PAROXETINE VIATRIS, en particulier si vous le faites brutalement, vous pouvez avoir des effets indésirables.
Dans la plupart des cas ces effets sont légers et disparaissent spontanément en 1 à 2 semaines. Parfois ces effets peuvent être plus sévères ou durer plus longtemps. Même si vous avez des effets lors du sevrage, vous pourrez quand même arrêter PAROXETINE VIATRIS.
A l'arrêt du traitement, les effets indésirables suivants peuvent survenir :
Des études ont montré que 3 personnes sur 10 ont présenté un ou plusieurs symptômes lors de l'arrêt de PAROXETINE VIATRIS, certains symptômes étant plus fréquents que d'autres.
Effets indésirables fréquents (moins de 1 personne sur 10) :
· vertiges (sensations vertigineuses, d'instabilité, perte d'équilibre)
· sensations de picotements, fourmillements, brûlures et plus rarement des sensations de décharges électriques dans la tête, bourdonnement, sifflement, sonnerie dans les oreilles (acouphènes),
· troubles du sommeil (rêves intenses, cauchemars, impossibilité de dormir),
· sensation d'anxiété,
· maux de tête.
Effets indésirables peu fréquents (moins de 1 personne sur 100) :
· nausées,
· transpiration excessive (y compris sueurs nocturnes),
· agitation, impatience des jambes,
· tremblements
· confusion (sentiment d'être confus ou désorienté),
· diarrhée,
· instabilité émotionnelle ou irritabilité,
· troubles visuels,
· palpitations.
Contactez votre médecin, si vous êtes inquiet(e) sur les effets liés à l’arrêt de PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables surviennent plus souvent dans les premières semaines de prise de PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable.
Si vous présentez un des effets indésirables suivants, contactez immédiatement votre médecin ou allez aux urgences.
Effets indésirables peu fréquents (moins de 1 personne sur 100) :
· saignement anormal (incluant vomissement de sang, sang dans les selles, ou « bleus »),
· difficulté ou impossibilité d’uriner.
Effets indésirables rares (moins de 1 personne sur 1000) :
· convulsions,
· agitation, impatiences des jambes, incapacité à rester assis ou debout sans bouger. Augmenter la dose de PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable pourrait aggraver ces sensations.
· fatigue, faiblesse, confusion, douleurs, raideurs musculaires ou mouvements involontaires des muscles (peut être en rapport avec un faible taux de sodium dans le sang).
Effets indésirables très rares (moins de 1 personne sur 10 000) :
· réactions allergiques à PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable qui peuvent être sévères : si vous présentez une rougeur ou des boursouflures au niveau de la peau, un gonflement des paupières, du visage, des lèvres, de la bouche ou de la langue, éruption cutanée ou urticaire n’importe où sur le corps, des démangeaisons, une difficulté pour respirer (essoufflement) ou pour avaler et une sensation de faiblesse ou d’étourdissement conduisant à un malaise ou une perte de connaissance, contactez immédiatement votre médecin ou allez aux urgences .
· Si vous avez certains ou tous les symptômes suivants, il se peut que vous ayez un syndrome sérotoninergique ou syndrome malin des neuroleptiques : les symptômes incluent une sensation de grande agitation ou d’irritabilité, de confusion, d’agitation, sensation de chaleur, une transpiration excessive, des tremblements, des frissons, des hallucinations (sons ou visions étranges), une rigidité des muscles, des myoclonies (secousses brusques des muscles), ou des battements du cœur rapides. La sévérité de ces symptômes peut s’aggraver conduisant à une perte de connaissance. Si vous ressentez cela contactez votre médecin.
· glaucome aigu.
Si vous avez une douleur oculaire et que votre vision devient trouble, contactez votre médecin.
Fréquence indéterminée
La fréquence ne peut être estimée avec les données disponibles.
· Des cas d’idées ou de comportements suicidaires ont été rapportés durant le traitement par PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ou peu après son arrêt (voir Idées suicidaires et aggravation de votre dépression ou de votre trouble anxieux à la rubrique 2. ).
· Des cas d’agression ont été observés durant le traitement par PAROXETINE VIATRIS. Si vous présentez ces effets indésirables, contactez votre médecin.
· Saignements vaginaux abondants peu après la naissance (hémorragie du post-partum), voir Grossesse, allaitement et fertilité à la rubrique 2 pour plus d'informations.
Autres effets indésirables possibles pendant le traitement
Effets indésirables très fréquents (plus de 1 personne sur 10) :
· nausées. La prise de votre médicament le matin pendant le petit-déjeuner réduit les risques de survenue de cet effet
· troubles sexuels. Par exemple, absence d’orgasme, et chez l’homme, érection et éjaculation anormales.
Effets indésirables fréquents (moins de 1 personne sur 10) :
· augmentation de la quantité de cholestérol dans le sang
· manque d’appétit
· troubles du sommeil : insomnie ou somnolence
· rêves anormaux (y compris cauchemars)
· sensations de vertiges ou de tremblements
· maux de tête
· difficultés à se concentrer
· agitation
· sensation de faiblesse
· vision floue
· bâillements
· bouche sèche
· diarrhée ou constipation
· vomissements
· prise de poids
· sueurs.
Effets indésirables peu fréquents (moins de 1 personne sur 100) :
· augmentation transitoire de la pression artérielle, ou baisse transitoire lors du passage rapide de la position assise à la position debout avec sensation vertigineuse ou de faiblesse
· accélération des battements du cœur
· absence de mouvements, rigidité, tremblements ou mouvements anormaux de la bouche et de la langue
· pupilles dilatées
· éruption cutanée
· démangeaisons
· confusion mentale
· hallucinations (visions ou sons étranges)
· émission involontaire et incontrôlable d’urine (incontinence urinaire) ou impossibilité d’uriner (rétention urinaire)
· si vous êtes diabétique, vous pouvez remarquer un déséquilibre du taux de sucre dans votre sang pendant la prise de Paroxétine Viatris 20 mg, comprimé pelliculé sécable. Parlez-en à votre médecin pour adapter la dose d’insuline ou des traitements antidiabétiques.
· diminution du nombre de globules blancs.
Effets indésirables rares (moins de 1 personne sur 1000) :
· manie (excitation, euphorie, )
· anxiété
· sensation d’être détaché de soi-même (dépersonnalisation)
· attaques de panique
· irrésistible envie de bouger les jambes (syndrome des jambes sans repos)
· battements lents du cœur
· élévation des valeurs de la fonction hépatique
· écoulement anormal de lait chez l’homme et la femme
· douleur dans les articulations ou les muscles
· augmentation dans le sang de l’hormone appelée prolactine
· troubles menstruels (incluant règles abondantes ou irrégulières, saignements en dehors des règles, et absence ou retard de règles).
Effets indésirables très rares (moins de 1 personne sur 10 000) :
· éruption cutanée, pouvant être accompagnée de cloques, et ressembler à de petites cibles (tâches centrales foncées bordées par une zone plus claire, et entourées d’un anneau foncé) appelée érythème polymorphe
· éruption cutanée généralisée accompagnée de cloques et d’un décollement de la peau, en particulier autour de la bouche, du nez, des yeux et des parties génitales (syndrome de Stevens-Johnson)
· éruption cutanée étendue accompagnée de cloques et d’un décollement de la peau sur une grande partie du corps (nécrolyse épidermique toxique ou syndrome de Lyell)
· atteinte hépatique pouvant entraîner une jaunisse au niveau de la peau et des yeux
· syndrome de sécrétion inappropriée de l’hormone anti-diurétique (SIADH) qui est un état dans lequel le corps présente un excès d’eau et une diminution de la concentration en sodium (sel), en raison de signaux chimiques inadaptés. Les patients présentant un SIADH peuvent être sévèrement malades, ou peuvent ne présenter aucun symptôme
· intolérance au soleil (photosensibilisation)
· rétention d’eau pouvant provoquer un œdème des bras et des jambes
· érection persistante et douloureuse du pénis
· diminution du nombre de plaquettes dans le sang.
Fréquence indéterminée
La fréquence ne peut être estimée avec les données disponibles
· Inflammation du colon (causant des diarrhées)
· Grincement des dents
· Quelques patients ont rapporté des sensations de bourdonnement, de sifflement ou de sonnerie dans les oreilles pendant le traitement par PAROXETINE VIATRIS. Une augmentation du risque de fractures osseuses a été observée chez des patients qui utilisent ce type de médicaments.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte, les plaquettes, le pilulier. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 25°C.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient PAROXETINE VIATRIS 20 mg, comprimé pelliculé sécable
· La substance active est :
Paroxétine base ......................................................................................................... 20,00 mg
Sous forme de chlorhydrate de paroxétine anhydre
Pour un comprimé pelliculé sécable.
· Les autres composants sont :
Hydrogénophosphate de calcium anhydre, carboxyméthylamidon sodique (type A), silice colloïdale anhydre, stéarate de magnésium.
Pelliculage : talc, dioxyde de titane (E171), copolymère de méthacrylate de butyle, de (2-diméthylaminoéthyl) méthacrylate et de méthylméthacrylate 1: 2: 1 (EUDRAGIT E 100).
Ce médicament se présente sous forme de comprimé pelliculé sécable.
Flacon de 14, 28, 30, 50, 100 ou 200 comprimés ou boîte de 14, 15, 28, 30, 50 ou 90 comprimés.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
1 RUE DE TURIN
69007 LYON
Exploitant de l’autorisation de mise sur le marché
1 RUE DE TURIN
69007 LYON
STATION CLOSE, POTTERS BAR
HERTFORDSHIRE - EN6 1TL
ROYAUME UNI
ou
VIATRIS SANTE
1 RUE DE TURIN
69007 LYON
ou
MYLAN HUNGARY LTD
MYLAN UTCA 1
2900 KOMAROM
HONGRIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).