Dernière mise à jour le 01/06/2026
ANETENA 3 mg/mL, collyre en solution en récipient unidose
Indications thérapeutiques
Classe pharmacothérapeutique : Anti-infectieux, antibiotiques, Code ATC : S01AA23.
ANETENA contient la substance active nétilmicine, un antibiotique qui tue les bactéries.
Il est indiqué chez les adultes pour le traitement local des infections externes de l’œil et de la région entourant l’œil causées par des bactéries sensibles à la nétilmicine.
Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien.
Présentations
> 15 flacon(s) unidose(s) (3x5) polyéthylène basse densité (PEBD) suremballée(s)/surpochée(s) de 0,3 ml
Code CIP : 34009 301 947 1 3
Déclaration de commercialisation : 06/12/2024
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 27/01/2023
ANETENA 3 mg/mL, collyre en solution en récipient unidose
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
1 mL de solution contient 3 mg de nétilmicine (sous forme de sulfate de nétilmicine).
Pour la liste complète des excipients, voir rubrique 6.1.
Collyre en solution en récipient unidose.
Solution limpide, incolore ou jaune pâle, pratiquement sans particules.
pH : 6,5 à 7,5
Osmolalité : 0,274 à 0,306 Osmol/kg
4.1. Indications thérapeutiques
Il convient de tenir compte des recommandations officielles concernant l’utilisation appropriée des antibactériens.
4.2. Posologie et mode d'administration
Une ou deux goutte(s) instillée(s) dans le cul-de-sac conjonctival de l’œil ou des yeux à traiter trois par jour ou conformément à la prescription médicale.
Population pédiatrique
La sécurité et l’efficacité de ANETENA 3 mg/mL collyre en solution chez les enfants (âgés de moins de 12 ans) et les adolescents (jusqu’à 18 ans) n’ont pas été établies.
Mode d’administration
|
1) Se laver soigneusement les mains avant d’instiller le collyre. |
|
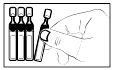
2) Ouvrir le sachet en aluminium contenant les récipients unidose. |
|
3) Vérifier que le récipient unidose est intact. |
|
|
|
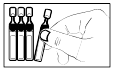
4) Détacher un récipient unidose de la bande et replacer les récipients non ouverts dans le sachet. |
|
|
|
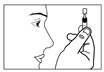
5) Ouvrir en tournant le capuchon du récipient sans tirer. Ne pas toucher l’embout après avoir ouvert le récipient. |
|
|
|
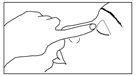
6) Presser doucement le récipient unidose de façon à ce qu’une seule goutte entre dans l’œil à traiter. Ne pas laisser l’embout du récipient unidose toucher l’œil ou la paupière ou toute autre surface afin d’éviter une possible contamination. |
|
|
|
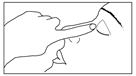
L’absorption systémique peut être réduite en comprimant le sac lacrymal au niveau du canthus interne pendant l’instillation du collyre et pendant une minute après l’instillation (cela bloque l’écoulement du collyre dans le canal lacrymo-nasal jusqu’à la surface absorbante étendue de la muqueuse naso-pharyngée). |
|
|


Durée du traitement
La durée habituelle de traitement est de 5 jours. Le médecin pourra prescrire un traitement plus long en cas d’infections réfractaires ou compliquées.
4.4. Mises en garde spéciales et précautions d'emploi
L’utilisation prolongée d’antibiotiques topiques peut entraîner une prolifération de microorganismes résistants. En l’absence d’amélioration clinique dans un délai relativement court ou en cas d’apparition d’une irritation ou d’une sensibilisation, le traitement doit être arrêté et un traitement approprié doit être instauré.
ANETENA n’est pas injectable ; par conséquent, il ne doit pas être administré par voie sous-conjonctivale ou introduit dans la chambre antérieure.
L’utilisation de lentilles de contact pendant une infection oculaire superficielle est fortement déconseillée.
Population pédiatrique
L’utilisation de ANETENA chez les enfants et adolescents n’est pas recommandée (voir rubrique 4.2).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L’administration concomitante d’autres antibiotiques potentiellement néphrotoxiques et ototoxiques, même topique et en particulier par voie intracavitaire, peut majorer le risque de tels effets.
L’administration concomitante ou séquentielle des médicaments néphrotoxiques ci-dessous avec des aminosides peut majorer le risque de néphrotoxicité et l’utilisation concomitante doit être évitée : cisplatine, polymyxine B, colistine, viomycine, streptomycine, vancomycine, autres aminosides et certaines céphalosporines (céphaloridine) ou diurétiques puissants tels que l’acide éthacrynique et le furosémide en raison de leurs effets sur les reins.
In vitro, l’association d’un aminoside et d’une bêta-lactamine (pénicillines ou céphalosporines) peut entraîner une inactivation réciproque et pertinente. Une diminution de la demi-vie ou de la concentration plasmatique de l’aminoside a été observée chez des patients atteints d’insuffisance rénale et chez certains patients ayant une fonction rénale normale, même lorsque l’aminoside et l’antibiotique de type pénicilline étaient administrés par deux voies différentes.
Les patients doivent être informés qu’en cas d’utilisation de plusieurs médicaments ophtalmiques, ceux-ci doivent être administrés à 5 minutes d’intervalle au moins. Les pommades doivent être appliquées en dernier.
4.6. Fertilité, grossesse et allaitement
Grossesse
Bien que les études précliniques n’aient pas mis en évidence de toxicité fœtale avec l’administration topique de nétilmicine en raison de la faible absorption systémique du produit, le médicament ne doit être administré pendant la grossesse qu’après une évaluation attentive du rapport bénéfice-risque et sous surveillance médicale étroite.
L’administration de ANETENA pendant l’allaitement n’est pas recommandée car les aminosides sont excrétés en faibles quantités dans le lait maternel.
Fertilité
Il n’existe pas de données concernant l’effet de ANETENA sur la fertilité humaine.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables rapportés sont présentés ci-dessous par classe de systèmes d’organes MedDRA. Les données disponibles ne sont pas suffisantes pour permettre de déterminer la fréquence de chacun des effets indésirables mentionnés (fréquence indéterminée).
Affections oculaires :
· irritation oculaire ;
· hyperémie conjonctivale ;
· rash de la paupière ;
· œdème palpébral ;
· prurit oculaire.
Affections du système immunitaire :
· hypersensibilité ;
· urticaire.
Les épisodes d’irritation oculaire et d’hypersensibilité causées par ANETENA sont bénins et transitoires.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Il n’a jamais été rapporté de cas de surdosage.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Anti-infectieux, antibiotiques, Code ATC : S01AA23.
La nétilmicine est un antibiotique aminoside semi-synthétique à large spectre. Elle est efficace à de faibles concentrations contre différentes bactéries pathogènes à Gram positif et à Gram négatif, y compris des souches résistantes à la gentamicine. Contrairement à la gentamicine, cet antibiotique n’est pas sensible à l’action inactivante des enzymes bactériennes qui catalysent les réactions de phosphorylation et de nucléotidylation.
La nétilmicine a un effet bactéricide rapide par induction d’erreurs de traduction dans le code génétique de l’ARNm, ce qui entraîne l’incorporation d’acides aminés incorrects dans la chaîne polypeptidique en élongation.
La prévalence de la résistance peut varier selon les régions et au cours du temps pour certaines espèces et des informations locales sur la résistance sont nécessaires, en particulier lors du traitement d’infections sévères. Les informations ci-dessous ne fournissent qu’une orientation approximative sur la probabilité qu’une bactérie soit sensible à la nétilmicine contenue dans ANETENA.
Les définitions des concentrations critiques classifiant les isolats comme sensibles ou résistants sont utiles pour prédire l’efficacité clinique des antibiotiques administrés par voie systémique. Cependant, lorsque l’antibiotique est administré par voie topique à des concentrations très élevées directement sur le site de l’infection, les définitions des concentrations critiques peuvent ne pas s’appliquer. Les infections par la plupart des isolats qui seraient classés comme résistants selon les concentrations critiques pour un antibiotique administré par voie systémique sont traitées efficacement par voie topique.
La fréquence de résistance globale aux aminosides peut aller jusqu’à 50 % de tous les staphylocoques dans certains pays européens.
Tableau 1 Valeurs seuils des CMI cliniques de nétilmicine en fonction des espèces (EUCAST 2017)
|
Microorganisme |
Valeurs seuils des CMI cliniques (mg/L) |
||
|
S (≤) |
R (≥) |
ECOFF |
|
|
Enterobacteriaceae |
2 |
4 |
2 |
|
Pseudomonas |
4 |
4 |
4 |
|
Acinetobacter |
4 |
4 |
NR |
|
Staphylococcus |
1 |
1 |
1 |
|
Staphylocoques à coagulase négative |
1 |
1 |
NR |
|
Enterococcus |
DI |
DI |
NR |
|
Streptococcus A, B, C et G |
NR |
NR |
NR |
|
Streptococcus pneumoniae |
NR |
NR |
NR |
|
Streptocoques viridans |
NR |
NR |
NR |
|
Haemophilus influenzae |
DI |
DI |
NR |
|
Moraxella catarrhalis |
DI |
DI |
NR |
|
Neisseria gonorrhea |
NR |
NR |
NR |
|
Neisseria meningitidis |
NR |
NR |
NR |
|
Bactéries anaérobies à Gram positif à l’exception de Clostridium difficile |
NR |
NR |
NR |
|
Bactéries anaérobies à Gram négatif |
NR |
NR |
NR |
|
Concentration critique non liée à l’espèce |
2 |
4 |
NR |
|
Notes : S = sensible. R = résistant. ECOFF = seuil épidémiologique harmonisé pour la surveillance de la résistance. DI = données insuffisante pour établir que l’espèce en question est une bonne cible pour un traitement avec ce médicament. NR = non rapportée. |
|||
Les études in vitro ont montré que la nétilmicine est active contre la plupart des souches des pathogènes oculaires courants et des bactéries de la flore cutanée courantes. Le tableau 2 présente une liste des niveaux de sensibilité à la nétilmicine pour un total de 767 isolats bactériens provenant d’échantillons oculaires cliniques prélevés en France (FR), Allemagne (DE), Italie (IT), Pologne (PL), Slovaquie (SK), Espagne (ES) et au Royaume-Uni (UK), et montre le niveau global de sensibilité de la flore oculaire courante à l’antibiotique.
Tableau 2 Données fréquentes de sensibilité à la nétilmicine in vitro issues d’isolats européens
|
|
Sensible |
Intermédiaire |
Résistant |
CMI50 (µg/mL) |
CMI90 (µg/mL) |
|||
|
Microorganisme |
[n] |
[%] |
[n] |
[%] |
[n] |
[%] |
||
|
S. aureus |
252 |
100 |
0 |
0 |
0 |
0 |
0,25 |
0,5 |
|
S. aureus (à coagulase négative) |
302 |
96,5 |
10 |
3,2 |
1 |
0,3 |
0,06 |
4 |
|
S. epidermidis |
216 |
95,6 |
9 |
4 |
1 |
0,4 |
0,05 |
4 |
|
S. pneumoniae |
|
|
|
|
|
|
4 |
8 |
|
H. influenzae |
|
|
|
|
|
|
0,25 |
0,5 |
|
P. aeruginosa |
39 |
100 |
0 |
0 |
0 |
0 |
4 |
4 |
Autres informations :
La résistance croisée entre les aminosides (par exemple gentamicine, tobramycine et nétilmicine) est due à la spécificité des enzymes modificatrices nucléotidyltransférases (ANT) et acétyltransférases (ACC). Cependant, la résistance croisée diffère entre les antibiotiques aminosides en raison de la spécificité variable des différentes enzymes modificatrices. Le mécanisme de résistance acquise aux aminosides le plus fréquent est l’inactivation de l’antibiotique par des enzymes modificatrices dont les gènes sont portés par des plasmides et des transposons.
5.2. Propriétés pharmacocinétiques
Après injection intramusculaire d’une dose de 2 mg/mL, une concentration plasmatique maximale de 5 µg/mL de nétilmicine est atteinte dans les 30 à 60 minutes. Après une perfusion intraveineuse de 60 minutes, la concentration plasmatique maximale est d’environ 11 µg/mL. La demi-vie est généralement de 2 à 2,5 heures chez les adultes et est prolongée en cas d’insuffisance rénale.
5.3. Données de sécurité préclinique
La DL50 par voie intramusculaire et intrapéritonéale est respectivement de 142 mg/kg et 186 mg/kg chez la souris et de 166 mg/kg et 266 mg/kg chez le rat ; chez le chien, la DL50 par voie IM est > 160 mg/kg et < 200 mg/kg et la DL50 par voie IV est > 40 mg/kg et < 72 mg/kg.
Hydroxyde de sodium (pour l’ajustement du pH)
Eau purifiée.
Récipients unidoses non ouverts : 24 mois.
Les récipients unidoses doivent être utilisés immédiatement après ouverture ; toute solution restante doit être éliminée.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25 °C.
Conserver les récipients unidoses dans le sachet en aluminium d'origine à l’abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
15 récipients unidoses en polyéthylène basse densité contenant chacun 0,3 mL de collyre en solution.
20 récipients unidoses en polyéthylène basse densité contenant chacun 0,3 mL de collyre en solution.
Chaque bande de 5 récipients unidoses est emballée dans un sachet en aluminium.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
VIA ERCOLE PATTI, 36
95025 ACI S. ANTONIO (CT)
ITALIE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 947 1 3 : 0,3 mL en flacon unidose (PEBD). Boîte de 15.
· 34009 550 701 5 3 : 0,3 mL en flacon unidose (PEBD). Boîte de 20.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 27/01/2023
ANETENA 3 mg/mL, collyre en solution en récipient unidose
nétilmicine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ANETENA 3 ml/mL, collyre en solution en récipient unidose et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser ANETENA 3 ml/mL, collyre en solution en récipient unidose?
3. Comment utiliser ANETENA 3 ml/mL, collyre en solution en récipient unidose?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ANETENA 3 ml/mL, collyre en solution en récipient unidose?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ANETENA 3 mg/mL, collyre en solution en récipient unidose ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Anti-infectieux, antibiotiques, Code ATC : S01AA23.
ANETENA contient la substance active nétilmicine, un antibiotique qui tue les bactéries.
Il est indiqué chez les adultes pour le traitement local des infections externes de l’œil et de la région entourant l’œil causées par des bactéries sensibles à la nétilmicine.
Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ANETENA 3 mg/mL, collyre en solution en récipient unidose?
· si vous êtes allergique à la nétilmicine, à tout autre antibiotique de la famille des aminosides (gentamicine, tobramycine, kanamycine, etc.) ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez vous à votre médecin ou pharmacien avant d’utiliser ANETENA.
En cas d’utilisation d’antibiotiques pendant une longue période, vous pourriez être plus prédisposé(e) aux infections oculaires.
Les antibiotiques de la famille des aminosides peuvent provoquer des troubles de l’audition et des troubles rénaux graves. Si vous utilisez un autre traitement antibiotique, y compris par voie orale, avec ANETENA, demandez conseil à votre médecin.
ANETENA n’est pas injectable ; par conséquent, le médicament ne doit pas être injecté dans l’œil ni introduit dans la partie avant de l’œil (« chambre antérieure »).
Dans tous les cas ci-dessus ou si vous présentez des réactions allergiques à ce médicament, arrêtez de l’utiliser et consultez votre médecin.
Enfants et adolescents
La sécurité et l’efficacité de ANETENA chez les enfants ou adolescents n’ont pas été établies.
Autres médicaments et ANETENA
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
ANETENA peut être utilisé avec d’autres médicaments ophtalmiques, mais il est important de suivre les instructions figurant à la rubrique 3.
Informez votre médecin ou votre pharmacien si vous prenez :
· tout autre antibiotique, en particulier : polymyxine B, colistine, viomycine, streptomycine, vancomycine ou céfaloridine. L’utilisation simultanée de ANETENA avec d’autres antibiotiques de la classe des aminosides pourrait augmenter le risque de troubles rénaux, de troubles de l’audition ou diminuer l’efficacité de l’autre antibiotique ;
· cisplatine (médicament anticancéreux) ;
· diurétiques (médicaments qui diminuent la rétention d’eau) tels que l’acide éthacrynique et le furosémide.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
Grossesse
Ce médicament ne doit être utilisé que si votre médecin juge que cela est approprié.
Allaitement
L’utilisation de ANETENA pendant la grossesse n’est pas recommandée car ce médicament est excrété en faibles quantités dans le lait maternel.
Conduite de véhicules et utilisation de machines
Votre vision peut être floue temporairement après l’instillation de ANETENA. Vous ne devez pas conduire ni utiliser des machines tant que votre vision n’est pas redevenue claire.
3. COMMENT UTILISER ANETENA 3 mg/mL, collyre en solution en récipient unidose?
La dose recommandée est d’une ou deux gouttes dans l’œil à traiter trois fois par jour ou conformément aux instructions de votre médecin.
Si vous portez des lentilles de contact
Le port de lentilles de contact pendant le traitement d’une infection oculaire superficielle est déconseillé.
Si vous portez des lentilles de contact, vous devez les retirer avant d’instiller les gouttes de ANETENA en récipients unidose car il ne contient pas de conservateur.
Mode d’emploi
1) Lavez-vous soigneusement les mains avant d’instiller le collyre.
2) Ouvrez le sachet en aluminium contenant les récipients unidoses.
|
3) Vérifier que le récipient unidose est intact. |
|
|
|
4) Détacher un récipient unidose de la bande et replacer les récipients non ouverts dans le sachet. |
|
|
|
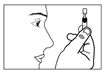
5) Ouvrir en tournant le capuchon du récipient sans tirer. 6) Ne pas toucher l’embout après avoir ouvert le récipient. |
|
|
|
7) Presser doucement le récipient unidose de façon à ce qu’une seule goutte entre dans l’œil à traiter. Ne pas laisser l’embout du récipient unidose toucher l’œil ou la paupière ou toute autre surface afin d’éviter une possible contamination. |
|
|
|
Gardez l’œil traité fermé et appuyez avec le bout du doigt sur le coin interne de l’œil fermé pendant une minute. C’est important car cela diminue la quantité de médicament qui passe dans le reste de l’organisme. |
|
|


Si vous avez utilisé plus de ANETENA que vous n’auriez dû
Il n’a jamais été rapporté de cas de surdosage lors de l’utilisation de ANETENA.
Si vous avez instillé accidentellement plus de gouttes que vous n’auriez dû, il est peu probable que cela provoque des problèmes. Instillez la dose suivante au moment habituel.
Si vous oubliez d’utiliser ANETENA
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez d’utiliser ANETENA
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
Si vous utilisez ANETENA avec d’autres médicaments ophtalmiques
Si vous utilisez un autre médicament ophtalmique, vous devez respecter un intervalle de 5 minutes entre l’administration de chaque médicament.
Les pommades ophtalmiques doivent être appliquées en dernier.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables suivants sont survenus avec ANETENA. La fréquence est indéterminée (ne peut être estimée sur la base des données disponibles) :
· irritation oculaire ;
· rougeur de l’œil ;
· éruption sur la paupière ;
· gonflement de la paupière ;
· démangeaisons de l’œil ;
· réaction allergique ;
· urticaire.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ANETENA 3 mg/mL, collyre en solution en récipient unidose ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et la boîte après « EXP ». La date de péremption fait référence au dernier jour de ce mois pour le produit non ouvert et conservé correctement.
A conserver à une température ne dépassant pas 25°C.
Conserver les récipients unidoses dans le sachet en aluminium d'origine à l’abri de la lumière.
Les gouttes sont présentées en récipients unidoses à usage unique. Après ouverture d’un récipient unidose, utiliser immédiatement et éliminer le récipient unidose après utilisation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est la nétilmicine. 1 mL de solution contient 3 mg de nétilmicine (sous forme de sulfate de nétilmicine).
· Les autres composants sont : chlorure de sodium, hydroxyde de sodium (pour l’ajustement du pH), eau purifiée.
Qu’est-ce que ANETENA et contenu de l’emballage extérieur
ANETENA est une solution limpide et incolore ou jaune pâle présentée en récipients unidoses qui sont emballés dans des sachets en aluminium conditionnés dans une boîte en carton.
Chaque boîte contient 15 ou 20 récipients unidoses.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
VIA ERCOLE PATTI, 36
95025 ACI S. ANTONIO (CT)
ITALIE
Exploitant de l’autorisation de mise sur le marché
8 RUE DES GRANDES TERRES
92500 RUEIL MALMAISON
FRANCE
VIA ERCOLE PATTI, 36
I-95025 ACI S. ANTONIO (CATANIA)
ITALIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
[1] Indiquer le libellé détaillé qui figure dans l’application form avec le code de la modification selon les lignes directrices https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-2/c_2013_2008/c_2013_2008_pdf/c_2013_2804_fr.pdf