Dernière mise à jour le 03/08/2026
RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
Indications thérapeutiques
RIVAROXABAN ZENTIVA vous a été prescrit parce que
· un syndrome coronarien aigu vous a été diagnostiqué (un ensemble de maladies incluant crise cardiaque et angor instable, correspondant à un type de douleur thoracique sévère) et parce que votre prise de sang a montré une augmentation des enzymes cardiaques.
Chez l’adulte, RIVAROXABAN ZENTIVA réduit le risque de présenter à nouveau une crise cardiaque ou de décéder des suites d’une maladie liée au cœur ou aux vaisseaux sanguins.
RIVAROXABAN ZENTIVA vous sera prescrit avec un autre médicament. Ainsi, votre médecin vous demandera également de prendre :
o de l’acide acétylsalicylique ou
o de l’acide acétylsalicylique plus du clopidogrel ou de la ticlopidine.
ou
· un risque élevé de présenter un caillot sanguin en raison d’une maladie coronarienne ou d’une maladie artérielle périphérique qui provoque des symptômes vous a été diagnostiqué.
Chez l’adulte, RIVAROXABAN ZENTIVA réduit le risque de présenter des caillots sanguins (événements athérothrombotiques).
RIVAROXABAN ZENTIVA vous sera prescrit avec un autre médicament. Ainsi, votre médecin vous demandera également de prendre de l’acide acétylsalicylique.
Dans certains cas, si vous recevez RIVAROXABAN ZENTIVA après une procédure consistant à désobstruer une artère rétrécie ou obstruée dans votre jambe pour restaurer la circulation sanguine, votre médecin pourra également vous prescrire du clopidogrel à prendre en plus de l’acide acétylsalicylique pendant une courte durée.
RIVAROXABAN ZENTIVA contient une substance active appelée rivaroxaban et appartient à une classe de médicaments appelés antithrombotiques. Il agit en bloquant un facteur de la coagulation sanguine (le facteur Xa), réduisant ainsi la tendance du sang à former des caillots.
Présentations
> plaquette(s) PVC aluminium de 56 comprimé(s)
Code CIP : 34009 301 984 4 5
Déclaration de commercialisation : 29/07/2024
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 12,37 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 13,39 €
- Taux de remboursement :15%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 21/03/2024
RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Rivaroxaban.......................................................................................................................... 2,5 mg
Pour un comprimé pelliculé.
Excipient à effet notoire :
Chaque comprimé pelliculé contient environ 27 mg de lactose (monohydraté).
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé.
Comprimé pelliculé jaune, rond, biconvexe (d’environ 5 mm de diamètre) comportant la mention « 2,5 » gravée en relief sur une face, l’autre face étant lisse.
4.1. Indications thérapeutiques
RIVAROXABAN ZENTIVA, co-administré avec de l’acide acétylsalicylique (AAS) seul ou avec de l’AAS plus du clopidogrel ou de la ticlopidine, est indiqué pour la prévention des événements athérothrombotiques chez les patients adultes suite à un syndrome coronarien aigu (SCA) avec élévation des biomarqueurs cardiaques (voir rubriques 4.3, 4.4 et 5.1).
RIVAROXABAN ZENTIVA, co-administré avec de l’acide acétylsalicylique (AAS), est indiqué pour la prévention des événements athérothrombotiques chez les patients adultes présentant une maladie coronarienne (MC) ou une maladie artérielle périphérique (MAP) symptomatique à haut risque d’événements ischémiques.
4.2. Posologie et mode d'administration
La dose recommandée est de deux prises par jour de 2,5 mg.
· SCA
Les patients sous RIVAROXABAN ZENTIVA 2,5 mg deux fois par jour doivent également prendre une dose quotidienne de 75-100 mg d’AAS ou une dose quotidienne de 75– 100 mg d’AAS en complément d’une dose quotidienne de 75 mg de clopidogrel ou d’une dose quotidienne standard de ticlopidine.
L’intérêt du traitement doit être régulièrement évalué au cas par cas après évaluation du risque d’événements ischémiques par rapport au risque de saignement. L’expérience étant limitée à 24 mois, une prolongation du traitement au-delà de 12 mois doit être définie au cas par cas (voir rubrique 5.1).
Le traitement par RIVAROXABAN ZENTIVA doit être débuté dès que possible après la phase de stabilisation du SCA (comprenant également les procédures de revascularisation) ; au plus tôt 24 heures après l’admission à l’hôpital et au moment où le patient ne requiert plus de traitement anticoagulant dans le cadre du SCA.
· MC/MAP
Les patients sous RIVAROXABAN ZENTIVA 2,5 mg deux fois par jour doivent également prendre une dose quotidienne de 75– 100 mg d’AAS.
Chez les patients ayant bénéficié d’une procédure de revascularisation réussie (chirurgicale ou endovasculaire, procédures hybrides incluses) d’un membre inférieur suite à une MAP symptomatique, le traitement ne doit pas être instauré avant que l’hémostase soit obtenue (voir rubrique 5.1).
La durée du traitement sera déterminée au cas par cas pour chaque patient de façon régulière et elle tiendra compte du risque d’événements thrombotiques par rapport au risque de saignements.
· SCA, MC/MAP
Administration concomitante avec un traitement antiplaquettaire
Chez les patients présentant un événement thrombotique aigu ou ayant subi une procédure vasculaire et nécessitant une bithérapie antiplaquettaire, la poursuite de RIVAROXABAN ZENTIVA 2,5 mg deux fois par jour devra être évaluée en fonction du type d’événement ou de procédure et du schéma posologique antiplaquettaire.
La sécurité et l’efficacité du rivaroxaban 2,5 mg deux fois par jour en association avec une bithérapie antiplaquettaire ont été étudiées chez des patients :
· ayant récemment présenté un SCA, en association avec l’ASS plus clopidogrel/ticlopidine (voir rubrique 4.1), et
· ayant récemment bénéficié d’une procédure de revascularisation d’un membre inférieur suite à une MAP symptomatique, en association avec l’AAS et, s’il y a lieu, avec du clopidogrel sur une courte durée (voir rubriques 4.4 et 5.1)
Oubli d’une dose
En cas d’oubli d’une dose, le patient doit poursuivre le traitement normalement en prenant la dose recommandée suivante à l’heure habituelle. La dose ne doit pas être doublée pour compenser une dose oubliée.
Relais des anti-vitamine K (AVK) par RIVAROXABAN ZENTIVA
Lors du passage des AVK à RIVAROXABAN ZENTIVA, les valeurs du rapport international normalisé (INR) pourraient être faussement élevées suite à la prise de RIVAROXABAN ZENTIVA. L’INR ne convient pas pour mesurer l’activité anticoagulante de RIVAROXABAN ZENTIVA et ne doit donc pas être utilisé (voir rubrique 4.5).
Relais de RIVAROXABAN ZENTIVA par les anti-vitamine K (AVK)
Il existe un risque d’anticoagulation inadéquate lors du relais de RIVAROXABAN ZENTIVA par les AVK. Une anticoagulation continue adéquate doit être assurée lors du relais par un autre anticoagulant. Il est à noter que RIVAROXABAN ZENTIVA peut contribuer à l’élévation de l’INR.
En cas de relais de RIVAROXABAN ZENTIVA par un AVK, l’AVK doit être administré conjointement jusqu’à ce que l’INR soit ≥ 2,0. Lors des deux premiers jours du relais, l’AVK doit être utilisé à sa posologie initiale standard, puis la posologie doit être adaptée sur la base des mesures de l’INR. Lorsque les patients reçoivent simultanément RIVAROXABAN ZENTIVA et l’AVK, l’INR doit être mesuré à partir de 24 heures après la dernière dose de RIVAROXABAN ZENTIVA et avant la dose suivante. Une fois le traitement par RIVAROXABAN ZENTIVA interrompu, des mesures fiables de l’INR ne peuvent être obtenues que 24 heures après la dernière dose de RIVAROXABAN ZENTIVA (voir rubriques 4.5 et 5.2).
Relais des anticoagulants parentéraux par RIVAROXABAN ZENTIVA
Chez les patients recevant un anticoagulant parentéral, arrêtez l’anticoagulant parentéral et initiez le traitement par RIVAROXABAN ZENTIVA 0 à 2 heures avant l’heure à laquelle l’administration suivante du médicament parentéral (héparines de bas poids moléculaire, par ex.) aurait été prévue ou au moment de l’arrêt du médicament parentéral en cas d’administration continue (héparine non fractionnée intraveineuse, par ex.).
Relais de RIVAROXABAN ZENTIVA par les anticoagulants parentéraux
La première dose d’anticoagulant parentéral doit être administrée à l’heure à laquelle la dose suivante de RIVAROXABAN ZENTIVA aurait dû être prise.
Populations particulières
Insuffisance rénale
Chez les patients atteints d’insuffisance rénale sévère (clairance de la créatinine de 15 à 29 mL/min), les données cliniques sont limitées mais montrent une augmentation significative des concentrations plasmatiques du rivaroxaban. Chez ces patients, RIVAROXABAN ZENTIVA doit donc être utilisé avec prudence. L'utilisation n’est pas recommandée chez les patients dont la clairance de la créatinine est < 15 mL/min (voir rubriques 4.4 et 5.2).
Aucun ajustement posologique n’est nécessaire chez les patients atteints d’insuffisance rénale légère (clairance de la créatinine de 50 à 80 mL/min) ou d’insuffisance rénale modérée (clairance de la créatinine de 30 à 49 mL/min) (voir rubrique 5.2).
Insuffisance hépatique
L’utilisation de RIVAROXABAN ZENTIVA est contre-indiquée chez les patients présentant une atteinte hépatique associée à une coagulopathie et à un risque de saignement cliniquement significatif, y compris chez les patients cirrhotiques avec un score de Child Pugh classe B ou C (voir rubriques 4.3 et 5.2).
Personnes âgées
Aucun ajustement posologique (voir rubriques 4.4 et 5.2).
Le risque de saignement augmente avec l’âge (voir rubrique 4.4).
Poids
Aucun ajustement posologique (voir rubriques 4.4 et 5.2).
Sexe
Aucun ajustement posologique (voir rubrique 5.2).
Population pédiatrique
La sécurité et l’efficacité du rivaroxaban 2,5 mg comprimé chez les enfants âgés de 0 à 18 ans n’ont pas été établies. Aucune donnée n’est disponible. L’utilisation des comprimés de RIVAROXABAN ZENTIVA 2,5 mg n’est donc pas recommandée chez l'enfant de moins de 18 ans.
Mode d’administration
RIVAROXABAN ZENTIVA est pour usage par voie orale.
Les comprimés peuvent être pris au cours ou en dehors des repas (voir rubriques 4.5 et 5.2).
Ecrasement des comprimés
Pour les patients qui sont dans l’incapacité d’avaler les comprimés entiers, le comprimé de RIVAROXABAN ZENTIVA peut être écrasé et mélangé à de l’eau ou de la compote de pommes, immédiatement avant utilisation pour être administré par voie orale.
Le comprimé écrasé peut également être administré au moyen d’une sonde gastrique (voir rubriques 5.2 et 6.6).
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Saignement évolutif cliniquement significatif.
Lésion ou maladie, si considérée comme étant à risque significatif de saignement majeur. Cela peut comprendre : ulcération gastro-intestinale en cours ou récente, présence de tumeurs malignes à haut risque de saignement, lésion cérébrale ou rachidienne récente, chirurgie cérébrale, rachidienne ou ophtalmique récente, hémorragie intracrânienne récente, varices œsophagiennes connues ou suspectées, malformations artérioveineuses, anévrismes vasculaires ou anomalies vasculaires majeures intrarachidiennes ou intracérébrales.
Traitement concomitant avec tout autre anticoagulant ; par exemple, héparine non-fractionnée (HNF), héparines de bas poids moléculaire (énoxaparine, daltéparine, etc), dérivés de l’héparine (fondaparinux, etc), anticoagulants oraux (warfarine, dabigatran etexilate, apixaban, etc) sauf dans des circonstances spécifiques de relais de traitement anticoagulant (voir rubrique 4.2) ou en cas d’administration d’HNF aux doses nécessaires pour le maintien de la perméabilité d’un cathéter central veineux ou artériel (voir rubrique 4.5).
Traitement concomitant du SCA avec un traitement antiplaquettaire chez les patients présentant des antécédents d’accident vasculaire cérébral (AVC) ou d’accident ischémique transitoire (AIT) (voir rubrique 4.4).
Traitement concomitant de la MC/MAP par de l’AAS chez les patients ayant déjà présenté un AVC hémorragique ou lacunaire, ou tout autre type d’AVC au cours du mois précédent (voir rubrique 4.4).
Atteinte hépatique associée à une coagulopathie et à un risque de saignement cliniquement significatif, y compris les patients cirrhotiques avec un score de Child Pugh classe B ou C (voir rubrique 5.2).
Grossesse et allaitement (voir rubrique 4.6).
4.4. Mises en garde spéciales et précautions d'emploi
Chez les patients présentant un SCA, l’efficacité et la sécurité du rivaroxaban 2,5 mg deux fois par jour ont été évaluées en association avec les agents antiplaquettaires, l’AAS seul ou l’AAS plus clopidrogrel/ticlopidine.
Chez les patients à haut risque d’événements ischémiques présentant une MC/MAP, l’efficacité et la sécurité du rivaroxaban 2,5 mg deux fois par jour ont été évaluées en association avec l’AAS.
Chez les patients ayant récemment bénéficié d’une procédure de revascularisation d’un membre inférieur suite à une MAP symptomatique, l’efficacité et la sécurité du rivaroxaban 2,5 mg deux fois par jour ont été évaluées en association avec l’agent antiplaquettaire AAS seul ou avec l’AAS plus clopidogrel utilisé sur une courte durée. Si elle est nécessaire, la bithérapie antiplaquettaire avec le clopidogrel doit être de courte durée ; une bithérapie antiplaquettaire de longue durée doit être évitée (voir rubrique 5.1).
Ce traitement en association avec d’autres agents antiplaquettaires, comme par ex. le prasugrel ou le ticagrelor, n’a pas été étudié et n’est donc pas recommandé.
Comme pour tout traitement anticoagulant, une surveillance clinique est recommandée pendant toute la durée du traitement.
Risque hémorragique
Comme avec les autres anticoagulants, les patients traités par RIVAROXABAN ZENTIVA doivent être surveillés étroitement à la recherche de tout signe de saignement. RIVAROXABAN ZENTIVA doit être utilisé avec prudence dans les situations présentant un risque hémorragique accru. Le traitement par RIVAROXABAN ZENTIVA doit être interrompu en cas d’hémorragie sévère (voir rubrique 4.9).
Au cours des études cliniques, des saignements des muqueuses (c.-à-d., épistaxis, saignement gingival, gastro-intestinal, génito-urinaire, dont des saignements vaginaux anormaux ou une augmentation des saignements menstruels) et des anémies ont été observés de manière plus fréquente durant le traitement au long cours par rivaroxaban associé à une mono ou bithérapie antiplaquettaire. Si nécessaire, des dosages de l’hémoglobine/des mesures de l’hématocrite pourraient permettre de détecter un saignement occulte et d’évaluer la pertinence clinique d’un saignement manifeste, en complément d’une surveillance clinique appropriée.
Plusieurs sous-groupes de patients, comme détaillés ci-dessous, présentent un risque majoré de saignement. Par conséquent, l’utilisation de RIVAROXABAN ZENTIVA en association avec une bithérapie antiplaquettaire chez les patients présentant une majoration connue du risque de saignement doit être évaluée au regard des bénéfices en termes de prévention des événements athérothrombotiques. En outre, ces patients doivent être surveillés attentivement à la recherche de signes et de symptômes de complications hémorragiques et d’anémie après l’instauration du traitement (voir rubrique 4.8).
Toute chute inexpliquée du taux d’hémoglobine ou de la pression artérielle doit amener à rechercher la présence de saignement.
Bien que le traitement par rivaroxaban ne nécessite pas de surveillance biologique de routine, la mesure des concentrations plasmatiques du rivaroxaban à l’aide de tests quantitatifs anti-facteur Xa étalonnés peut être utile dans des situations exceptionnelles pour lesquelles la connaissance de l’exposition au rivaroxaban peut aider à la décision clinique, comme dans le cas d’un surdosage ou d’une intervention chirurgicale en urgence (voir rubriques 5.1 et 5.2).
Insuffisance rénale
En cas d’insuffisance rénale sévère (clairance de la créatinine < 30 mL/min), les concentrations plasmatiques du rivaroxaban peuvent être augmentées de manière significative (d’un facteur 1,6 en moyenne), ce qui peut majorer le risque de saignement. RIVAROXABAN ZENTIVA doit être utilisé avec prudence chez les patients dont la clairance de la créatinine est comprise entre 15 et 29 mL/min. L’utilisation de RIVAROXABAN ZENTIVA n’est pas recommandée chez les patients dont la clairance de la créatinine est < 15 mL/min (voir rubriques 4.2 et 5.2).
Chez les patients atteints d’insuffisance rénale modérée (clairance de la créatinine de 30 à 49 mL/min) recevant de façon concomitante d’autres médicaments augmentant les concentrations plasmatiques du rivaroxaban, RIVAROXABAN ZENTIVA doit être utilisé avec prudence (voir rubrique 4.5).
Interaction avec d’autres médicaments
L’utilisation de RIVAROXABAN ZENTIVA n’est pas recommandée chez les patients recevant simultanément un traitement systémique par un antifongique azolé (tel que le kétoconazole, l’itraconazole, le voriconazole et le posaconazole) ou un inhibiteur de la protéase du VIH (ritonavir, par ex.). Ces substances actives sont de puissants inhibiteurs du CYP3A4 et de la glycoprotéine P (P-gp) et peuvent donc augmenter les concentrations plasmatiques du rivaroxaban à un niveau cliniquement significatif (d’un facteur 2,6 en moyenne), ce qui peut majorer le risque de saignement (voir rubrique 4.5).
Une attention particulière est nécessaire chez les patients traités simultanément par des médicaments modifiant l’hémostase, tels que les anti-inflammatoires non stéroïdiens (AINS), l’acide acétylsalicylique (AAS) et les antiagrégants plaquettaires ou les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS) et les inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSN). Chez les patients à risque de maladie ulcéreuse gastro-intestinale, un traitement prophylactique approprié peut être envisagé (voir rubriques 4.5 et 5.1).
Les patients traités par RIVAROXABAN ZENTIVA et des agents antiplaquettaires ne doivent recevoir un traitement concomitant par des AINS que si le bénéfice prévaut sur le risque de saignement.
Autres facteurs de risque hémorragique
Comme les autres médicaments antithrombotiques, le rivaroxaban n’est pas recommandé chez les patients présentant un risque accru de saignement, notamment dans les cas suivants :
· syndromes hémorragiques congénitaux ou acquis,
· hypertension artérielle sévère non contrôlée,
· maladie gastro-intestinale sans ulcération active pouvant potentiellement entraîner des complications hémorragiques (par ex. maladie inflammatoire chronique des intestins, œsophagite, gastrite et reflux gastro-œsophagien),
· rétinopathie vasculaire,
· bronchectasie ou antécédents de saignement pulmonaire.
Il doit être utilisé avec prudence chez les patients présentant un SCA et une MC/MAP :
· chez les patients âgés de ≥ 75 ans lorsqu’il est co-administré avec de l’AAS seul ou avec de l’AAS plus clopidogrel ou ticlopidine. Le rapport bénéfice/risque du traitement doit être évalué au cas par cas, de façon régulière ;
· ayant un faible poids corporel (< 60 kg) lorsqu’il est co-administré avec de l’AAS seul ou avec de l’AAS plus clopidogrel ou ticlopidine ;
· chez les patients présentant une MC avec insuffisance cardiaque symptomatique sévère. Les données de l’étude indiquent que ces patients pourraient tirer moins de bénéfice avec un traitement par rivaroxaban (voir rubrique 5.1).
Patients atteints de cancer
Les patients atteints d'une maladie maligne peuvent présenter simultanément un risque plus élevé de saignements et de thrombose. Le bénéfice individuel du traitement antithrombotique doit être évalué par rapport au risque de saignement chez les patients atteints d'un cancer actif, en fonction de la localisation de la tumeur, du traitement antinéoplasique et du stade de la maladie. Les tumeurs localisées dans les voies gastrointestinales ou génito-urinaires ont été associées à un risque accru de saignements pendant le traitement par le rivaroxaban.
Chez les patients atteints de néoplasmes malins à haut risque de saignements, l'utilisation du rivaroxaban est contre-indiquée (voir rubrique 4.3).
Patients porteurs de valves artificielles
Le rivaroxaban ne doit pas être utilisé dans le cadre d’une thromboprophylaxie chez les patients ayant subi récemment un remplacement de valve aortique par voie transcathéter (RVAT). La sécurité et l’efficacité du rivaroxaban n’ont pas été étudiées chez les patients porteurs de prothèses valvulaires cardiaques ; aucune donnée ne permet donc d’établir que le rivaroxaban puisse maintenir une anticoagulation appropriée chez cette population de patients. L’utilisation de RIVAROXABAN ZENTIVA n’est pas recommandée chez ces patients.
Patients présentant un syndrome des antiphospholipides
Les anticoagulants oraux à action directe (AOD) incluant le rivaroxaban ne sont pas recommandés chez les patients présentant des antécédents de thrombose chez lesquels a été diagnostiqué un syndrome des antiphospholipides. En particulier pour les patients positifs aux trois tests antiphospholipides (anticoagulant circulant lupique, anticorps anticardiolipine et anticorps anti-bêta 2-glycoprotéine I), le traitement par AOD pourrait être associé à des taux plus élevés de récidives d’événements thrombotiques que ceux observés en cas de traitement par un antagoniste de la vitamine K.
Patients présentant des antécédents d’AVC et/ou d’AIT
Patients présentant un SCA
L’utilisation de RIVAROXABAN ZENTIVA 2,5 mg est contre-indiquée pour le traitement d’un SCA chez les patients présentant des antécédents d’AVC ou d’AIT (voir rubrique 4.3). Les données d’efficacité chez les patients traités suite à un SCA et présentant des antécédents d’AVC ou d’AIT sont limitées mais indiquent que le traitement n’apporte pas de bénéfice chez ces patients.
Patients présentant une MC/MAP
Les patients présentant une MC/MAP et ayant des antécédents d’accident vasculaire cérébral hémorragique ou lacunaire ou d’accident vasculaire cérébral ischémique non lacunaire au cours du mois précédent n’ont pas été étudiés (voir rubrique 4.3).
Les patients ayant récemment bénéficié de procédures de revascularisation d’un membre inférieur suite à une MAP symptomatique présentant également des antécédents d’AVC ou d’AIT n’ont pas été étudiés. Le traitement par RIVAROXABAN ZENTIVA 2,5 mg doit être évité chez ces patients recevant une bithérapie antiplaquettaire.
Anesthésie péridurale/rachidienne ou ponction péridurale/lombaire
La réalisation d’anesthésie rachidienne/péridurale ou de ponction lombaire/péridurale chez les patients traités par des médicaments antithrombotiques en prévention de complications thrombo-emboliques entraîne un risque d’apparition d’un hématome péridural ou rachidien pouvant provoquer une paralysie prolongée ou permanente. Ce risque peut être majoré par l’utilisation postopératoire de cathéters périduraux à demeure ou par l’utilisation concomitante de médicaments modifiant l’hémostase. Le risque peut également être augmenté en cas de ponction lombaire ou péridurale répétée ou traumatique. Les patients doivent faire l’objet d’une surveillance fréquente avec recherche de signes et symptômes d’atteinte neurologique (par ex., engourdissement ou faiblesse des jambes, dysfonctionnement des intestins ou de la vessie). Si des troubles neurologiques apparaissent, il est nécessaire de réaliser un diagnostic et un traitement de toute urgence. Avant toute intervention cérébrospinale, le médecin doit évaluer les bénéfices potentiels ainsi que le risque encouru chez les patients sous anticoagulants ou chez les patients devant être placés sous anticoagulants en vue d’une prévention antithrombotique. Il n’y a pas d’expérience clinique de l’utilisation du rivaroxaban 2,5 mg avec des agents antiplaquettaires dans ces situations. Les anti-agrégants plaquettaires doivent être arrêtés comme préconisé dans les résumés des caractéristiques du produit de ces médicaments.
Afin de réduire le risque potentiel de saignement lors de la réalisation d’une anesthésie rachidienne/péridurale ou d’une ponction lombaire chez les patients recevant un traitement par rivaroxaban, le profil pharmacocinétique du rivaroxaban doit être pris en compte. Il est préférable de réaliser la pose ou le retrait d’un cathéter péridural ou une ponction lombaire lorsque l’effet anticoagulant du rivaroxaban est estimé faible (voir rubrique 5.2). Cependant, le délai précis pour atteindre un effet anticoagulant suffisamment faible chez chaque patient n’est pas connu.
Recommandations posologiques avant et après des gestes invasifs et interventions chirurgicales
Si un geste invasif ou une intervention chirurgicale est requise, le traitement par RIVAROXABAN ZENTIVA 2,5 mg doit être interrompu au moins 12 heures avant l’intervention si possible, et doit reposer sur l’appréciation clinique du médecin. Si un patient doit faire l’objet d’une intervention chirurgicale programmée et si l’effet antiplaquettaire n’est pas souhaité, l’administration d’antiagrégants plaquettaires doit être interrompue comme indiqué dans le Résumé des Caractéristiques du Produit des médicaments concernés.
Si le geste ne peut être différé, la majoration du risque hémorragique doit être évaluée au regard de l’urgence de l’intervention.
Le traitement par RIVAROXABAN ZENTIVA doit être réinstauré dès que possible après le geste invasif ou l’intervention chirurgicale à condition que la situation clinique le permette et qu’une hémostase adéquate ait pu être obtenue, telle que déterminée par le médecin (voir rubrique 5.2).
Personnes âgées
Le risque hémorragique peut augmenter avec l’âge (voir rubriques 5.1 et 5.2).
Réactions cutanées
Pendant la période de surveillance post-commercialisation du rivaroxaban, des réactions cutanées graves, incluant des syndromes de Stevens-Johnson/nécrolyse épidermique toxique et des syndromes de réaction d’hypersensibilité médicamenteuse avec éruptions cutanées généralisées, fièvre élevée, éosinophilie et atteintes systémiques (syndrome DRESS), ont été signalées lors de l’utilisation du rivaroxaban (voir rubrique 4.8). Le risque d’apparition de ces réactions chez les patients semble être plus élevé en début de traitement : dans la majorité des cas, la réaction survient dans les premières semaines de traitement. Le rivaroxaban doit être arrêté dès la première apparition d’une éruption cutanée sévère (par ex : une éruption diffuse, intense et/ou bulleuse) ou de tout autre signe d’hypersensibilité accompagné de lésions des muqueuses.
Excipients
RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Inhibiteurs du CYP3A4 et de la P-gp
L’administration concomitante de rivaroxaban et de kétoconazole (400 mg une fois par jour) a entraîné une augmentation de 2,6 fois la valeur moyenne de l’ASC et de 1,7 fois la valeur moyenne de la Cmax du rivaroxaban, avec une majoration significative des effets pharmacodynamiques du rivaroxaban ; de même, l’administration concomitante de rivaroxaban et de ritonavir (600 mg deux fois par jour) a entraîné une augmentation de 2,5 fois la valeur moyenne de l’ASC et de 1,6 fois la valeur moyenne de la Cmax du rivaroxaban, avec une majoration significative des effets pharmacodynamiques du rivaroxaban. Ces associations peuvent entraîner un risque majoré de saignement. L’utilisation de RIVAROXABAN ZENTIVA n’est donc pas recommandée chez les patients recevant simultanément un traitement systémique par un antifongique azolé, tel que le kétoconazole, l’itraconazole, le voriconazole ou le posaconazole, ou un inhibiteur de la protéase du VIH. Ces substances actives sont de puissants inhibiteurs du CYP3A4 et de la P-gp (voir rubrique 4.4).
Les substances actives inhibant de manière importante une seule des voies d’élimination du rivaroxaban, à savoir le CYP3A4 ou la P-gp, devraient augmenter dans une moindre mesure les concentrations plasmatiques du rivaroxaban. Par exemple, l’administration de clarithromycine (500 mg deux fois par jour), considérée comme un puissant inhibiteur du CYP3A4 et un inhibiteur modéré de la P-gp, a entraîné une augmentation de 1,5 fois la valeur moyenne de l’ASC et de 1,4 fois la valeur moyenne de la Cmax du rivaroxaban L’interaction avec la clarithromycine semble ne pas être cliniquement pertinente chez la plupart des patients mais pourrait être potentiellement significative chez les patients à haut risque. (Pour les patients insuffisants rénaux, voir rubrique 4.4).
L’érythromycine (500 mg trois fois par jour), qui inhibe modérément le CYP3A4 et la P-gp, a entraîné une augmentation de 1,3 fois la valeur moyenne de l’ASC et de la Cmax moyenne du rivaroxaban. L’interaction avec l’érythromycine semble ne pas être cliniquement pertinente chez la plupart des patients mais pourrait être potentiellement significative chez les patients à haut risque.
Chez les sujets atteints d’insuffisance rénale légère, l’érythromycine (500 mg trois fois par jour) a entraîné une augmentation de 1,8 fois la valeur moyenne de l’ASC et une augmentation de 1,6 fois la valeur moyenne de la Cmax du rivaroxaban par comparaison aux sujets présentant une fonction rénale normale. Chez les sujets atteints d’insuffisance rénale modérée, l’érythromycine a entraîné une augmentation de 2,0 fois la valeur moyenne de l’ASC et une augmentation de 1,6 fois la valeur moyenne de la Cmax du rivaroxaban par comparaison aux sujets présentant une fonction rénale normale. L’effet de l’érythromycine s’additionne à celui de l’insuffisance rénale (voir rubrique 4.4).
Le fluconazole (400 mg une fois par jour), considéré comme un inhibiteur modéré du CYP3A4, a entraîné une augmentation de 1,4 fois la valeur moyenne de l’ASC et de 1,3 fois la valeur moyenne de la Cmax du rivaroxaban. L’interaction avec le fluconazole semble ne pas être cliniquement pertinente chez la plupart des patients mais pourrait être potentiellement significative chez les patients à haut risque. (Pour les patients insuffisants rénaux, voir rubrique 4.4).
Les données cliniques disponibles avec la dronédarone étant limitées, cette association doit donc être évitée.
Anticoagulants
Après administration concomitante d’énoxaparine (40 mg en dose unique) et de rivaroxaban (10 mg en dose unique), un effet additif sur l’activité anti-facteur Xa a été observé, sans effet supplémentaire sur les tests de coagulation (TQ, TCA). L’énoxaparine n’a pas eu d’incidence sur les caractéristiques pharmacocinétiques du rivaroxaban.
Compte tenu du risque accru de saignement, une prudence particulière est nécessaire en cas de traitement concomitant avec tout autre anticoagulant (voir rubriques 4.3 et 4.4).
AINS/antiagrégants plaquettaires
Aucun allongement cliniquement significatif du temps de saignement n’a été observé après administration concomitante de rivaroxaban (15 mg) et de naproxène 500 mg. La réponse pharmacodynamique peut néanmoins s’avérer plus marquée chez certaines personnes.
Aucune interaction pharmacocinétique ou pharmacodynamique cliniquement significative n’a été observée lors de l’administration concomitante de rivaroxaban et d’acide acétylsalicylique 500 mg.
Aucune interaction pharmacocinétique avec le rivaroxaban (15 mg) n’a été observée lors de l’utilisation de clopidogrel (dose de charge de 300 mg puis dose d’entretien de 75 mg), mais une augmentation significative du temps de saignement a été constatée dans un sous-groupe de patients sans corrélation avec les taux d’agrégation plaquettaire, de la P-sélectine ou du récepteur GPIIb/IIIa.
La prudence est nécessaire si les patients sont traités simultanément par des AINS (dont l’acide acétylsalicylique) ou des antiagrégants plaquettaires car ces médicaments augmentent habituellement le risque de saignement (voir rubrique 4.4).
ISRS/IRSN
Comme avec les autres anticoagulants, il est possible que les patients soient exposés à un risque accru de saignement en cas d’utilisation simultanée d’ISRS ou d’IRSN en raison des effets rapportés de ces médicaments sur les plaquettes. Lors de leur utilisation concomitante au cours du programme clinique du rivaroxaban, des taux numériquement supérieurs d’événements hémorragiques majeurs ou de saignements non majeurs cliniquement pertinents ont été observés dans tous les groupes de traitement.
Warfarine
Le passage de la warfarine, un anti-vitamine K (INR de 2,0 à 3,0) au rivaroxaban (20 mg) ou du rivaroxaban (20 mg) à la warfarine (INR de 2,0 à 3,0) a entraîné une augmentation du temps de Quick/INR (Néoplastine) au-delà d’un effet purement additif (des INR individuels allant jusqu’à 12 peuvent être observés), alors que les effets sur le TCA, sur l’inhibition de l’activité du facteur Xa et sur l’ETP (Endogenous Thrombin Potential) ont été additifs.
Si les effets pharmacodynamiques du rivaroxaban doivent être testés pendant la période de relais, l’activité anti-facteur Xa, le PiCT et le Heptest peuvent être utilisés, ces tests n’ayant pas été affectés par la warfarine. Dès le quatrième jour après la dernière dose de warfarine, tous les tests (y compris le TQ, le TCA, l’inhibition de l’activité du facteur Xa et l’ETP) ont reflété uniquement les effets du rivaroxaban.
Si les effets pharmacodynamiques de la warfarine doivent être testés pendant la période de relais, la mesure de l’INR peut être utilisée à la Cmin du rivaroxaban (24 heures après la prise précédente du rivaroxaban), ce test n’étant affecté que de façon minime par le rivaroxaban pendant cette période.
Aucune interaction pharmacocinétique n’a été observée entre la warfarine et le rivaroxaban.
Inducteurs du CYP3A4
L’administration concomitante de rivaroxaban et de rifampicine, un puissant inducteur du CYP3A4, a entraîné une diminution d’environ 50 % de l’ASC moyenne du rivaroxaban, associée à une réduction de ses effets pharmacodynamiques. L’utilisation concomitante de rivaroxaban et d’autres inducteurs puissants du CYP3A4 (phénytoïne, carbamazépine, phénobarbital ou millepertuis [Hypericum perforatum], par ex.) peut également entraîner une réduction des concentrations plasmatiques du rivaroxaban. En conséquence, les inducteurs puissants du CYP3A4 doivent être évités à moins que le patient ne bénéficie d’une surveillance étroite des signes et symptômes de thrombose.
Autres traitements concomitants
Aucune interaction pharmacocinétique ou pharmacodynamique cliniquement significative n’a été observée lors de l’administration concomitante de rivaroxaban et de midazolam (substrat du CYP 3A4), de digoxine (substrat de la P-gp), d’atorvastatine (substrat du CYP3A4 et de la P-gp) ou d’oméprazole (inhibiteur de la pompe à protons). Le rivaroxaban n’a pas d’effet inhibiteur ou inducteur sur les isoformes principaux du CYP, tels que le CYP3A4.
Aucune interaction cliniquement significative avec les aliments n’a été observée (voir rubrique 4.2).
Effets sur les analyses de laboratoire
Les valeurs des paramètres de la coagulation (TQ, TCA, Heptest, par ex.) sont modifiées comme le laisse prévoir le mode d’action du rivaroxaban (voir rubrique 5.1).
4.6. Fertilité, grossesse et allaitement
Grossesse
La sécurité et l’efficacité du rivaroxaban n’ont pas été établies chez la femme enceinte. Les études réalisées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Considérant le risque potentiel de toxicité sur la reproduction, le risque intrinsèque de saignement et le passage de la barrière placentaire par le rivaroxaban, RIVAROXABAN ZENTIVA est contre-indiqué pendant la grossesse (voir rubrique 4.3).
Les femmes en âge de procréer doivent éviter toute grossesse au cours du traitement par rivaroxaban.
La sécurité et l’efficacité du rivaroxaban n’ont pas été établies chez les mères allaitantes. Les données recueillies chez l’animal indiquent que le rivaroxaban passe dans le lait maternel. En conséquence, RIVAROXABAN ZENTIVA est contre-indiqué pendant l’allaitement (voir rubrique 4.3). Un choix doit donc être fait entre l’arrêt de l’allaitement ou l’interruption/la non prise de RIVAROXABAN ZENTIVA.
Fertilité
Aucune étude spécifique n’a été menée chez l’Homme pour évaluer les effets du rivaroxaban sur la fertilité. Aucun effet n’a été observé dans une étude sur la fertilité des mâles et des femelles chez le rat (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
La tolérance du rivaroxaban a été évaluée dans treize études pivots de phase III (voir tableau 1).
Au total, 69 608 patients adultes inclus dans 19 études de phase III et 488 patients pédiatriques inclus dans deux études de phase II et deux études de phase III ont été exposés au rivaroxaban.
Tableau 1 : Nombre de patients étudiés, dose quotidienne totale et durée maximale du traitement dans les études de phase III menées chez des patients adultes et pédiatriques
|
Indication |
Nombre de patients* |
Dose quotidienne totale |
Durée maximale du traitement |
|
Prévention des événements thromboemboliques veineux (ETEV) chez les patients adultes bénéficiant d’une chirurgie programmée de la hanche ou du genou |
6 097 |
10 mg |
39 jours |
|
Prévention des ETEV chez les patients présentant une affection médicale aiguë |
3 997 |
10 mg |
39 jours |
|
Traitement de la thrombose veineuse profonde (TVP), de l’embolie pulmonaire (EP) et prévention des récidives |
6 790 |
Jours 1– 21 : 30 mg Jour 22 et suivants : 20 mg Après au moins 6 mois : 10 mg ou 20 mg |
21 mois |
|
Traitement des ETEV et prévention des récidives sous forme d’ETEV chez les nouveau-nés nés à terme et chez les enfants âgés de moins de 18 ans après l’instauration d’un traitement anticoagulant standard |
329 |
Dose ajustée selon le poids corporel pour atteindre une exposition similaire à celle observée chez les adultes traités pour une TVP avec 20 mg de rivaroxaban une fois par jour |
12 mois |
|
Prévention des AVC et des embolies systémiques chez les patients atteints de fibrillation atriale non valvulaire |
7 750 |
20 mg |
41 mois |
|
Prévention des événements athérothrombotiques suite à un syndrome coronarien aigu (SCA) |
10 225 |
5 mg ou 10 mg respectivement, co-administrés avec de l’AAS ou de l’AAS associé au clopidogrel ou à la ticlopidine |
31 mois |
|
Prévention des événements athérothrombotiques chez les patients présentant une MC/MAP |
18 244 |
5 mg co-administrés avec de l’AAS ou 10 mg seuls |
47 mois |
|
3 256** |
5 mg co-administrés avec de l’AAS |
42 mois |
* Patients exposés à au moins une dose de rivaroxaban
** Dans l’étude VOYAGER PAD
Les effets indésirables signalés le plus fréquemment chez les patients recevant du rivaroxaban ont été les saignements (voir rubrique 4.4. et « Description de certains effets indésirables » ci-dessous) (tableau 2). Parmi les saignements signalés, les plus fréquents ont été l’épistaxis (4,5 %) et l’hémorragie du tractus gastro-intestinal (3,8 %).
Tableau 2 : Taux de survenue des saignements* et des anémies chez les patients exposés au rivaroxaban au cours des études de phase III terminées chez des patients adultes et pédiatriques
|
Indication |
Tout saignement |
Anémie |
|
Prévention des ETEV chez les patients adultes bénéficiant d’une chirurgie programmée de la hanche ou du genou |
6,8 % des patients |
5,9 % des patients |
|
Prévention des ETEV chez les patients présentant une affection médicale aiguë |
12,6 % des patients |
2,1 % des patients |
|
Traitement de la TVP, de l’EP et prévention des récidives |
23 % des patients |
1,6 % des patients |
|
Traitement des ETEV et prévention des récidives sous forme d’ETEV chez les nouveau-nés nés à terme et chez les enfants âgés de moins de 18 ans après l’instauration d’un traitement anticoagulant standard |
39,5 % des patients |
4,6 % des patients |
|
Prévention des AVC et des embolies systémiques chez les patients atteints de fibrillation atriale non valvulaire |
28 pour 100 patient-années |
2,5 pour 100 patient-années |
|
Prévention des événements athérothrombotiques suite à un SCA |
22 pour 100 patient-années |
1,4 pour 100 patient-années |
|
Prévention des événements athérothrombotiques chez les patients présentant une MC/MAP |
6,7 pour 100 patient-années |
0,15 pour 100 patient-années** |
|
8,38 pour 100 patient-années# |
0,74 pour 100 patient-années*** # |
* Pour toutes les études sur le rivaroxaban, tous les événements hémorragiques sont recueillis, rapportés et adjudiqués.
** Dans l’étude COMPASS, il y a une faible incidence des anémies car une approche sélective du recueil des événements indésirables a été utilisée
*** Une approche sélective du recueil des évènements indésirables a été utilisée
# Dans l’étude VOYAGER PAD
Tableau résumant les effets indésirables
Les fréquences des effets indésirables rapportés avec le rivaroxaban dans la population adulte et pédiatrique sont résumées dans le tableau 3 ci-dessous par classe de systèmes d’organes (classification MedDRA) et par fréquence.
Les fréquences sont définies comme suit :
· très fréquent (≥ 1/10)
· fréquent (≥ 1/100, < 1/10)
· peu fréquent (≥ 1/1 000, < 1/100)
· rare (≥ 1/10 000, < 1/1 000)
· très rare (< 1/10 000)
· fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
Tableau 3 : Ensemble des effets indésirables rapportés chez les patients adultes dans les essais cliniques de phase III ou par une utilisation post-commercialisation* ainsi que dans deux études pédiatriques de phase II et deux études pédiatriques de phase III
|
Fréquent |
Peu fréquent |
Rare |
Très rare |
Fréquence indéterminée |
|
Affections hématologiques et du système lymphatique |
||||
|
Anémie (dont résultat d’analyse de laboratoire correspondant) |
Thrombocytose (dont élévation de la numération plaquettaire)A, thrombopénie |
|
|
|
|
Affections du système immunitaire |
||||
|
|
Réaction allergique, dermatite allergique, œdème de Quincke et œdème allergique |
|
Réactions anaphylactiques, y compris choc anaphylactique |
|
|
Affections du système nerveux |
||||
|
Sensations vertigineuses, céphalées |
Hémorragie cérébrale et intracrânienne, syncope |
|
|
|
|
Affections oculaires |
||||
|
Hémorragie oculaire (dont hémorragie conjonctivale) |
|
|
|
|
|
Affections cardiaques |
||||
|
|
Tachycardie |
|
|
|
|
Affections vasculaires |
||||
|
Hypotension, hématomes |
|
|
|
|
|
Affections respiratoires, thoraciques et médiastinales |
||||
|
Épistaxis, hémoptysie |
|
|
Pneumonie à éosinophiles |
|
|
Affections gastro-intestinales |
||||
|
Gingivorragie, hémorragie du tractus gastro-intestinal (dont rectorragie), douleurs gastro-intestinales et abdominales, dyspepsie, nausées, constipationA, diarrhée, vomissementsA |
Sécheresse buccale |
|
|
|
|
Affections hépatobiliaires |
||||
|
Elévation des transaminases |
Insuffisance hépatique, élévation de la bilirubine, élévation des phosphatases alcalines sanguinesA, élévation des γ-GTA |
Ictère, élévation de la bilirubine conjuguée (avec ou sans élévation concomitante des ALAT), cholestase, hépatite (dont lésion hépatocellulaire) |
|
|
|
Affections de la peau et du tissu sous-cutané |
||||
|
Prurit (dont cas peu fréquents de prurit généralisé), éruption cutanée, ecchymose, hémorragie cutanée et sous-cutanée |
Urticaire |
|
Syndrome de Stevens-Johnson/Nécrolyse épidermique toxique, syndrome DRESS |
|
|
Affections musculo-squelettiques et systémiques |
||||
|
Douleur des extrémitésA |
Hémarthrose |
Hémorragie musculaire |
|
Syndrome de compression des loges secondaire à un saignement |
|
Affections du rein et des voies urinaires |
||||
|
Hémorragie du tractus urogénital (dont hématurie et ménorragieB), insuffisance rénale (dont élévation de la créatinine plasmatique, élévation de l’urée plasmatique) |
|
|
|
Insuffisance rénale/insuffisance rénale aiguë secondaire à un saignement suffisant pour provoquer une hypoperfusion, néphropathie liée aux anticoagulants |
|
Troubles généraux et anomalies au site d’administration |
||||
|
FièvreA, œdème périphérique, réduction générale de la vivacité (dont fatigue et asthénie) |
Sensation d’inconfort (dont malaise) |
Œdème localiséA |
|
|
|
Investigations |
||||
|
|
Élévation de la LDHA, de la lipaseA, de l’amylaseA |
|
|
|
|
Lésions, intoxications et complications liées aux procédures |
||||
|
Hémorragie post-opératoire (dont anémie postopératoire et hémorragie de la plaie), contusion, plaie suintanteA |
|
Pseudoanévrisme vasculaireC |
|
|
A : effets observés dans la prévention des ETEV chez les patients adultes bénéficiant d’une intervention chirurgicale programmée de la hanche ou du genou (prothèse totale de hanche ou du genou)
B : effets observés très fréquemment chez les femmes âgées de < 55 ans dans le traitement de la TVP, de l’EP et la prévention des récidives
C : effets observés peu fréquemment dans la prévention des événements athérothrombotiques suite à un SCA (suite à une intervention coronaire percutanée)
* Une approche sélective prédéfinie du recueil des événements indésirables a été utilisée dans certaines études de phase III. L’incidence des effets indésirables n’a pas augmenté et qu’aucun nouvel effet indésirable n’a été identifié à la suite de l’analyse de ces études.
Description de certains effets indésirables
En raison du mode d’action pharmacologique du produit, l’utilisation du rivaroxaban peut être associée à un risque accru de saignement occulte ou apparent au niveau de tout organe ou tissu, ce qui peut entraîner une anémie post-hémorragique. Les signes, les symptômes et la sévérité (y compris les évolutions fatales) dépendront de la localisation et du degré ou de l’étendue du saignement et/ou de l’anémie (voir rubrique 4.9 « Prise en charge des saignements »). Au cours des études cliniques, des saignements des muqueuses (c. -à-d. épistaxis, saignement gingival, gastro-intestinal, génito-urinaire, dont des saignements vaginaux anormaux ou une augmentation des saignements menstruels) et des anémies ont été observés de manière plus fréquente durant le traitement au long cours par rivaroxaban comparé au traitement par AVK. Si nécessaire, des dosages de l’hémoglobine/des mesures de l’hématocrite pourraient permettre de détecter un saignement occulte et d’évaluer la pertinence clinique d’un saignement manifeste, en complément d’une surveillance clinique appropriée. Le risque de saignement peut être augmenté chez certains groupes de patients, par ex. en cas d’hypertension artérielle sévère non contrôlée et/ou de traitement concomitant modifiant l’hémostase (voir rubrique 4.4 « Risque hémorragique »). Les saignements menstruels peuvent être amplifiés et/ou prolongés.
Des complications hémorragiques peuvent se manifester sous forme de faiblesse, de pâleur, de sensations vertigineuses, de céphalées ou de gonflements inexpliqués, de dyspnée et d’état de choc inexpliqué. Dans certains cas, en conséquence de l’anémie, des symptômes d’ischémie cardiaque tels qu’une douleur thoracique ou une angine de poitrine, ont été observés.
Des complications connues, secondaires à une hémorragie sévère, telles qu’un syndrome de compression des loges et une insuffisance rénale due à l’hypoperfusion, ou une néphropathie liée aux anticoagulants ont été signalées sous rivaroxaban. Par conséquent, l’éventualité d’une hémorragie doit être envisagée lors de l’évaluation de toute affection chez un patient sous anticoagulant.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
De rares cas de surdosage à des doses allant jusqu’à 1 960 mg ont été signalés. En cas de surdosage, le patient doit être surveillé attentivement pour détecter toute complication hémorragique ou autre effet indésirable (voir rubrique « Prise en charge des saignements »).. A des doses suprathérapeutiques de 50 mg ou plus de rivaroxaban, et en raison de l’absorption limitée du produit, un effet de plafonnement sans augmentation supplémentaire de l’exposition plasmatique moyenne est attendu.
Un agent de réversion spécifique (andexanet alfa) permettant de contrer les effets pharmacodynamiques du rivaroxaban est disponible (se référer au Résumé des Caractéristiques du Produit de l’andexanet alfa).
L’utilisation de charbon actif peut être envisagée afin de limiter l’absorption en cas de surdosage au rivaroxaban.
Prise en charge des saignements
En cas de survenue d’une complication à type de saignement chez un patient recevant du rivaroxaban, l’administration suivante du rivaroxaban doit être différée ou le traitement doit être interrompu, si nécessaire. La demi-vie du rivaroxaban est d’environ 5 à 13 heures (voir rubrique 5.2). La prise en charge doit être définie au cas par cas selon la sévérité et la localisation de l’hémorragie. Un traitement symptomatique adapté pourra être utilisé si besoin, tel que la compression mécanique (en cas d’épistaxis sévère, par ex.), le rétablissement chirurgical de l’hémostase avec contrôle du saignement, le remplissage vasculaire et la correction hémodynamique, les transfusions sanguines (concentrés de globules rouges ou plasma frais congelé, selon l’anémie ou la coagulopathie associée) ou plaquettaires.
Si les mesures ci-dessus ne suffisent pas à contrôler le saignement, l’administration soit d’un agent de réversion spécifique des inhibiteurs du facteur Xa (andexanet alfa), permettant de contrer les effets pharmacodynamiques du rivaroxaban, soit d’un agent procoagulant spécifique, par exemple un concentré de complexe prothrombinique (CCP), un concentré de complexe prothrombinique activé (CCPA) ou du facteur VIIa recombinant (r-FVIIa), doit être envisagée. A ce jour cependant, l’expérience clinique de l’utilisation de ces médicaments chez les personnes traitées par le rivaroxaban est très limitée. Cette recommandation est également basée sur des données non cliniques limitées. Un nouveau dosage et une adaptation de la dose du facteur VIIa recombinant doivent être envisagés en fonction de l’évolution du saignement. En fonction des disponibilités locales, une consultation avec un spécialiste de la coagulation doit être envisagée en cas de saignements majeurs (voir rubrique 5.1).
Aucun effet du sulfate de protamine ou de la vitamine K sur l’activité anticoagulante du rivaroxaban n’est attendu. Il n’existe que des données limitées sur l’utilisation de l’acide tranexamique et aucune donnée sur l’utilisation de l’acide aminocaproïque et de l’aprotinine chez les personnes traitées par le rivaroxaban. En outre, il n’existe pas de justification scientifique sur des bénéfices potentiels ni d’expérience sur l’utilisation de l’agent hémostatique systémique, la desmopressine, chez les personnes traitées par le rivaroxaban. Etant donné la forte liaison du rivaroxaban aux protéines plasmatiques, le produit n’est probablement pas dialysable.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le rivaroxaban est un inhibiteur direct hautement sélectif du facteur Xa, doté d’une biodisponibilité par voie orale. L’inhibition du facteur Xa interrompt les voies intrinsèque et extrinsèque de la cascade de coagulation sanguine, inhibant ainsi la formation de thrombine et le développement du thrombus.
Le rivaroxaban n’inhibe pas la thrombine (facteur II activé) et aucun effet sur les plaquettes n’a été démontré.
Effets pharmacodynamiques
Une inhibition dose-dépendante de l’activité du facteur Xa a été observée chez l’être humain. Le temps de Quick (TQ) est influencé par le rivaroxaban de façon dose-dépendante et étroitement liée à la concentration plasmatique en rivaroxaban (r = 0,98), lorsque la Néoplastine est utilisée comme réactif. Des résultats différents pourraient être observés avec d’autres réactifs. Le résultat du TQ doit être exprimé en secondes car l’INR est étalonné et validé uniquement pour les coumariniques et ne peut pas être utilisé avec les autres anticoagulants.
Une étude de pharmacologie clinique portant sur l’antagonisation des effets pharmacodynamiques du rivaroxaban chez des sujets adultes sains (n = 22) a évalué les effets de doses uniques (50 UI/kg) de deux types de CCP différents : un CCP contenant 3 facteurs (facteurs II, IX et X) et un CCP contenant 4 facteurs (facteurs II, VII, IX et X). Le CCP contenant 3 facteurs a entrainé une réduction des valeurs moyennes du TQ (Néoplastine) d’environ 1,0 seconde dans les 30 minutes, par rapport à des réductions d’environ 3,5 secondes observées avec le CCP contenant 4 facteurs. En revanche, le CCP contenant 3 facteurs a eu un effet global plus rapide et plus important d’antagonisation des modifications de la génération de thrombine endogène par rapport au CCP à 4 facteurs (voir rubrique 4.9).
Les valeurs du temps de céphaline activé (TCA) et du HepTest sont également allongées de manière dose-dépendante ; leur utilisation n’est toutefois pas recommandée pour évaluer les effets pharmacodynamiques du rivaroxaban. Il n’est pas nécessaire de surveiller en routine les paramètres de coagulation pendant le traitement par rivaroxaban. Cependant, si cela est cliniquement indiqué, les concentrations plasmatiques en rivaroxaban peuvent être mesurées à l’aide de tests quantitatifs anti-facteur Xa étalonnés (voir rubrique 5.2).
Efficacité et sécurité clinique
SCA
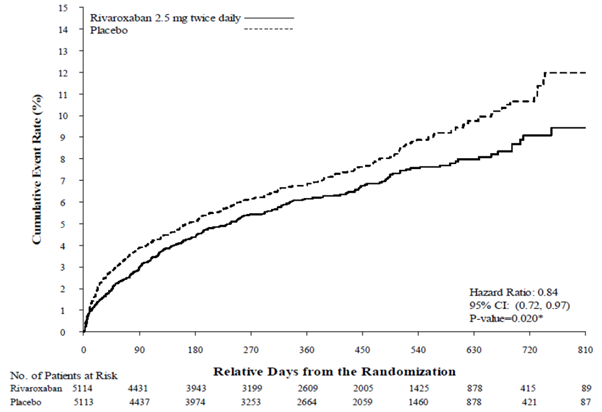
Le programme clinique du rivaroxaban a été conçu pour démontrer l’efficacité du rivaroxaban en prévention des décès d’origine cardiovasculaire (CV), des infarctus du myocarde (IDM) ou des AVC chez les patients ayant récemment présenté un SCA (IDM avec sus-décalage du segment ST [IM ST+], IM sans sus-décalage du segment ST [IM non ST+] ou angor instable [AI]). Lors de l’étude pivot en double aveugle ATLAS ACS 2 TIMI 51, 15 526 patients ont été randomisés selon un ratio de 1/1/1 dans trois groupes de traitement : rivaroxaban 2,5 mg par voie orale deux fois par jour, 5 mg par voie orale deux fois par jour ou placebo deux fois par jour co-administré avec AAS ou AAS plus une thiénopyridine (clopidogrel ou ticlopidine). Les patients présentant un SCA âgés de moins de 55 ans devaient être diabétiques ou avoir des antécédents d’IDM. La durée médiane de traitement a été de 13 mois et la durée totale de traitement a atteint presque 3 ans. 93,2 % des patients ont reçu simultanément de l’AAS plus un traitement par thiénopyridine et 6,8 % uniquement de l’AAS. Parmi les patients ayant reçu un double traitement antiplaquettaire, 98,8 % ont reçu du clopidogrel, 0,9 % ont reçu de la ticlopidine et 0,3 % ont reçu du prasugrel. Les patients ont reçu la première dose de rivaroxaban dès que possible après la phase de stabilisation du SCA, comprenant les procédures de revascularisation, au minimum 24 heures et jusqu’à 7 jours (moyenne : 4,7 jours) après leur admission à l’hôpital, et au moment où le patient ne requiert plus de traitement anticoagulant dans le cadre du SCA.
Les traitements par le rivaroxaban 2,5 mg deux fois par jour et le rivaroxaban 5 mg deux fois par jour ont tous deux été efficaces, en association à un traitement conventionnel par antiplaquettaires pour réduire davantage l’incidence des événements CV. Le traitement à 2,5 mg deux fois par jour a réduit la mortalité et il existe des données en faveur du fait que la dose la plus faible a été associée à un moindre risque de saignement ; par conséquent, le rivaroxaban 2,5 mg deux fois par jour, co-administré avec de l’acide acétylsalicylique (AAS) seul ou avec de l’AAS plus une thiénopyridine (clopidogrel ou ticlopidine) est recommandé pour la prévention des événements athérothrombotiques chez les patients adultes suite à un SCA avec élévation des biomarqueurs cardiaques.
Le rivaroxaban a significativement réduit l’incidence de survenue des événements du critère principal d’efficacité composite comprenant les décès d’origine CV, les IDM ou les AVC par comparaison avec le placebo. Le bénéfice s’est révélé par une diminution des décès d’origine CV et des IDM et est apparu précocement avec un effet constant du traitement pendant la totalité de la période de traitement (voir tableau 4 et figure 1). La fréquence de survenue du premier des critères secondaires d’efficacité (décès toutes causes confondues, IDM ou AVC) a également été réduite de façon significative.
Une analyse supplémentaire rétrospective a montré une réduction numérique significative du taux d’incidence des thromboses intra-stent par comparaison avec le placebo (voir tableau 4). Les taux d’incidence relevés pour le critère principal de tolérance (événements hémorragiques majeurs TIMI non associés à un pontage aorto-coronarien [PAC]) ont été plus élevés chez les patients traités par rivaroxaban que chez les patients ayant reçu le placebo (voir tableau 6). Cependant, les taux d’incidence des composantes du critère principal de tolérance, que sont les événements hémorragiques fatals, l’hypotension nécessitant un traitement par des agents inotropes intraveineux et les interventions chirurgicales pour hémorragies prolongées, ont été équilibrés entre le rivaroxaban et le placebo.
Les résultats d’efficacité des patients ayant bénéficié d’une intervention coronaire percutanée (ICP) sont présentés dans le tableau 5. Les résultats de tolérance dans ce sous-groupe de patients ayant bénéficié d’une ICP étaient comparables aux résultats dans la population générale.
Les patients avec élévation des biomarqueurs (troponine or CK-MB) et sans antécédent d’AVC/AIT ont constitué 80 % de la population étudiée. Les résultats dans cette population ont également été cohérents par rapport aux résultats d’efficacité et de tolérance dans la population générale.
Tableau 4 : Données d’efficacité de l’étude de phase III ATLAS ACS 2 TIMI 51
|
Population de l’étude |
Patients ayant présenté récemment un SCAa) |
|
|
Posologie |
Rivaroxaban 2,5 mg, deux fois par jour, N = 5 114 n (%) Hazard Ratio (HR) (IC à 95 %) valeur de pb) |
Placebo N = 5 113 n (%) |
|
Décès d’origine cardiovasculaire, IDM ou AVC |
313 (6,1 %) 0,84 (0,72-0,97) p = 0,020* |
376 (7,4 %) |
|
Décès toutes causes confondues, IDM ou AVC |
320 (6,3 %) 0,83 (0,72-0,97) p = 0,016* |
386 (7,5 %) |
|
Décès d’origine cardiovasculaire |
94 (1,8 %) 0,66 (0,51-0,86) p = 0,002** |
143 (2,8 %) |
|
Décès toutes causes confondues |
103 (2,0 %) 0,68 (0,53-0,87) p = 0,002** |
153 (3,0 %) |
|
IDM |
205 (4,0 %) 0,90 (0,75-1,09) p = 0,270 |
229 (4,5 %) |
|
AVC |
46 (0,9 %) 1,13 (0,74-1,73) p = 0,562 |
41 (0,8 %) |
|
Thrombose intra-stent |
61 (1,2 %) 0,70 (0,51-0,97) p = 0,033** |
87 (1,7 %) |
a) population en intention de traiter modifiée (population en intention de traiter pour thrombose intra-stent)
b) par comparaison avec le placebo ; valeur de p selon le test du Log-Rank
* statistiquement supérieur
** numériquement significatif
Tableau 5 : Données d’efficacité de l’étude de phase III ATLAS ACS 2 TIMI 51 chez les patients ayant bénéficié d’une ICP
|
Population de l’étude |
Patients ayant présenté récemment un SCA et ayant bénéficié d’une ICPa) |
|
|
Posologie |
Rivaroxaban 2,5 mg, deux fois par jour, N = 3 114 n (%) HR (IC à 95 %) valeur de pb) |
Placebo N = 3 096 n (%) |
|
Décès d’origine cardiovasculaire, IDM ou AVC |
153 (4,9 %) 0,94 (0,75-1,17) p = 0,572 |
165 (5,3 %) |
|
Décès d’origine cardiovasculaire |
24 (0,8 %) 0,54 (0,33-0,89) p = 0,013** |
45 (1,5 %) |
|
Décès toutes causes confondues |
31 (1,0 %) 0,64 (0,41-1,01) p = 0,053 |
49 (1,6 %) |
|
IDM |
115 (3,7 %) 1,03 (0,79-1,33) p = 0,829 |
113 (3,6 %) |
|
AVC |
27 (0,9 %) 1,30 (0,74-2,31) p = 0,360 |
21 (0,7 %) |
|
Thrombose intra-stent |
47 (1,5 %) 0,66 (0,46-0,95) p = 0,026** |
71 (2,3 %) |
a) population en intention de traiter modifiée (population en intention de traiter pour thrombose intra-stent)
b) par comparaison avec le placebo ; valeur de p selon le test du Log-Rank
** numériquement significatif
Tableau 6 : Données de tolérance de l’étude de phase III ATLAS ACS 2 TIMI 51
|
Population de l’étude |
Patients ayant présenté récemment un SCAa) |
|
|
Posologie |
Rivaroxaban 2,5 mg, deux fois par jour, N = 5 115 n (%) HR (IC à 95 %) valeur de pb) |
Placebo N = 5 125 n(%) |
|
Événement hémorragique majeur TIMI non associé à un pontage aorto-coronarien |
65 (1,3 %) 3,46 (2,08– 5,77) p = < 0,001* |
19 (0,4 %) |
|
Événement hémorragique fatal |
6 (0,1 %) 0,67 (0,24– 1,89) p = 0,450 |
9 (0,2 %) |
|
Hémorragie intracrânienne symptomatique |
14 (0,3 %) 2,83 (1,02– 7,86) p = 0,037 |
5 (0,1 %) |
|
Hypotension nécessitant un traitement par des agents inotropes intraveineux |
3 (0,1 %) |
3 (0,1 %) |
|
Intervention chirurgicale pour hémorragie prolongée |
7 (0,1 %) |
9 (0,2 %) |
|
Transfusion de 4 unités ou plus de sang sur une période de 48 heures |
19 (0,4 %) |
6 (0,1 %) |
a) population d’évaluation de la tolérance, sous traitement
b) par comparaison avec le placebo ; valeur de p selon le test du Log-Rank
* statistiquement significatif
Figure 1 : Délai avant première survenue du critère principal d’efficacité (décès d’origine CV, IDM ou AVC)

MC/MAP
L’étude de phase III COMPASS (27 395 patients, 78,0 % d’hommes, 22,0 % de femmes) a démontré l’efficacité et la sécurité du rivaroxaban dans la prévention du critère composite incluant décès CV, IDM, AVC chez les patients présentant une MC ou une MAP symptomatique à haut risque d’événements ischémiques. Les patients étaient suivis pendant une durée médiane de 23 mois et un maximum de 3,9 ans.
Les sujets ne nécessitant pas un traitement continu par un inhibiteur de la pompe à protons étaient randomisés au pantoprazole ou au placebo. Puis, tous les patients étaient randomisés selon un rapport 1/1/1 au rivaroxaban 2,5 mg deux fois par jour/AAS 100 mg une fois par jour, au rivaroxaban 5 mg deux fois par jour, ou à l’AAS 100 mg une fois par jour seul, et leurs placebos respectifs.
Les patients présentant une MC avaient une MC affectant plusieurs lits vasculaires et/ou un antécédent d’IDM. Pour les patients âgés de < 65 ans, il était nécessaire qu’ils présentent une athérosclérose impliquant au moins deux lits vasculaires ou au moins deux autres facteurs de risque cardiovasculaires.
Les patients présentant une MAP avaient des antécédents d’interventions, tels qu’un pontage ou une angioplastie transluminale percutanée ou l’amputation d’un membre ou d’un pied pour une maladie vasculaire artérielle ou une claudication intermittente avec un index de pression systolique cheville/bras < 0,90 et/ou une sténose artérielle périphérique significative ou une précédente revascularisation de la carotide ou une sténose de l'artère carotide asymptomatique ≥ 50 %.
Les critères de non inclusion incluaient les patients nécessitant une bithérapie antiplaquettaire ou un antiplaquettaire autre que l’AAS ou un traitement par anticoagulant oral et les patients à haut risque de saignement, ou présentant une insuffisance cardiaque avec une fraction d’éjection < 30 % ou une insuffisance cardiaque de classe III ou IV selon la New York Heart Association, ou tout accident vasculaire cérébral ischémique, non lacunaire, survenu dans le mois précédent ou tout antécédent d’accident vasculaire cérébral hémorragique ou lacunaire.
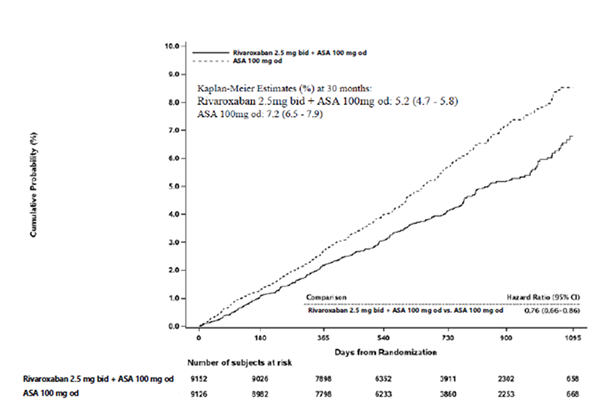
Le rivaroxaban 2,5 mg deux fois par jour en association avec de l’AAS 100 mg une fois par jour était supérieur à l’AAS 100 mg dans la réduction du critère composite principal incluant décès CV, IDM, AVC (voir tableau 7 et figure 2).
Une augmentation significative du critère principal de sécurité (événements hémorragiques majeurs selon la classification modifiée de l’ISTH) a été observée chez les patients traités par rivaroxaban 2,5 mg deux fois par jour en association avec de l’AAS 100 mg une fois par jour par rapport aux patients recevant de l’AAS 100 mg une fois par jour (voir tableau 8).
Pour le critère principal d’efficacité, le bénéfice observé du rivaroxaban 2,5 mg deux fois par jour en association avec de l’AAS 100 mg une fois par jour par rapport à l’AAS 100 mg une fois par jour était d’un HR de 0,89 (IC à 95 % : 0,7– 1,1) chez les patients âgés de ≥ 75 ans (incidence : 6,3 % vs 7,0 %) et d’un HR de 0,70 (IC à 95 % : 0,6– 0,8) chez les patients âgés de < 75 ans (3,6 % vs 5,0 %). Pour les hémorragies majeures selon la classification modifiée de l’ISTH, l’augmentation du risque observée était d’un HR de 2,12 (IC à 95 % : 1,5– 3,0) chez les patients âgés de ≥ 75 ans (5,2 % vs 2,5 %) et d’un HR de 1,53 (IC à 95 % : 1,2– 1,9) chez les patients âgés de < 75 ans (2,6 % vs 1,7 %).
Chez les patients ne nécessitant pas d'un inhibiteur de la pompe à protons sur le plan clinique, l'utilisation du pantoprazole 40 mg une fois par jour en association au médicament antithrombotique à l'étude n'a pas montré de bénéfice pour la prévention des événements gastro-intestinaux hauts (c.-à-d. hémorragie gastro-intestinale haute, ulcération gastro-intestinale haute ou obstruction ou perforation gastro-intestinale haute) ; le taux d'incidence des événements gastro-intestinaux hauts était de 0,39/100 années-patients dans le groupe pantoprazole 40 mg une fois par jour et 0,44/100 années-patients dans le groupe placebo une fois par jour.
Tableau 7 : Données d’efficacité de l’étude de phase III COMPASS
|
Population de l’étude |
Patients présentant une MC/MAPa) |
|
||||
|
Posologie |
Rivaroxaban 2,5 mg 2x/j en association avec de l’AAS 100 mg 1x/j N = 9 152 |
AAS 100 mg 1x/j
N = 9 126 |
|
|||
|
|
Patients avec événements |
KM % |
Patients avec événements |
KM % |
HR (IC à 95 %) |
Valeur de pb) |
|
|
||||||
|
AVC, IDM ou décès CV |
379 (4,1 %) |
5,20 % |
496 (5,4 %) |
7,17 % |
0,76 (0,66 ; 0,86) |
p = 0,00004* |
|
- AVC |
83 (0,9 %) |
1,17 % |
142 (1,6 %) |
2,23 % |
0,58 (0,44 ; 0,76) |
p = 0,00006 |
|
- IDM |
178 (1,9 %) |
2,46 % |
205 (2,2 %) |
2,94 % |
0,86 (0,70 ; 1,05) |
p = 0,14458 |
|
- Décès CV |
160 (1,7 %) |
2,19 % |
203 (2,2 %) |
2,88 % |
0,78 (0,64 ; 0,96) |
p = 0,02053 |
|
|
||||||
|
Mortalité toutes causes |
313 (3,4 %) |
4,50 % |
378 (4,1 %) |
5,57 % |
0,82 (0,71 ; 0,96) |
|
|
Ischémie aiguë des membres |
22 (0,2 %) |
0,27 % |
40 (0,4 %) |
0,60 % |
0,55 (0,32 ; 0,92) |
|
a) analyse sur la population en intention de traiter, analyses principales
b) vs AAS 100 mg ; valeur de p du Log-Rank
* La réduction pour le critère principal d’efficacité était statistiquement supérieure.
2x/j : deux fois par jour ; IC : intervalle de confiance ; KM % : estimation du risque d’incidence cumulée calculé à 900 jours selon Kaplan-Meier ; CV : cardiovasculaire ; IDM : infarctus du myocarde ; 1x/j : une fois par jour
Tableau 8 : Données de sécurité de l’étude de phase III COMPASS
|
Population de l’étude |
Patients présentant une MC/MAPa) |
||
|
Posologie |
Rivaroxaban 2,5 mg 2x/j en association avec de l’AAS 100 mg 1x/j, N = 9 152 n (Risque cum. %) |
AAS 100 mg 1x/j
N = 9 126 n (Risque cum. %) |
Hazard Ratio (IC à 95 %)
Valeur de pb) |
|
Hémorragies majeures selon la classification modifiée de l’ISTH |
288 (3,9 %)
|
170 (2,5 %) |
1,70 (1,40 ; 2,05) p < 0,00001 |
|
- Événement hémorragique fatal |
15 (0,2 %) |
10 (0,2 %) |
1,49 (0,67 ; 3,33) p = 0,32164 |
|
- Hémorragie symptomatique affectant un organe critique (non fatale) |
63 (0,9 %) |
49 (0,7 %) |
1,28 (0,88 ; 1,86) p = 0,19679 |
|
- Hémorragie au niveau d’un site chirurgical nécessitant une réopération (non fatale, pas dans un organe critique) |
10 (0,1 %) |
8 (0,1 %) |
1,24 (0,49 ; 3,14) p = 0,65119 |
|
- Hémorragie conduisant à une hospitalisation (non fatale, pas dans un organe critique, ne nécessitant pas une réopération) |
208 (2,9 %) |
109 (1,6 %) |
1,91 (1,51 ; 2,41) p < 0,00001 |
|
Avec séjour d’une nuit |
172 (2,3 %) |
90 (1,3 %) |
1,91 (1,48 ; 2,46) p < 0,00001 |
|
Sans séjour d’une nuit |
36 (0,5 %) |
21 (0,3 %) |
1,70 (0,99 ; 2,92) p = 0,04983 |
|
- Hémorragie gastro-intestinale majeure |
140 (2,0 %) |
65 (1,1 %) |
2,15 (1,60 ; 2,89) p < 0,00001 |
|
- Hémorragie intra-crânienne majeure |
28 (0,4 %) |
24 (0,3 %) |
1,16 (0,67 ; 2,00) p = 0,59858 |
a) analyse sur la population en intention de traiter, analyses principales
b) vs AAS 100 mg ; valeur de p du Log-Rank
2x/j. : deux fois par jour ; IC : intervalle de confiance ; Risque cum. : risque de l’incidence cumulée (estimations de Kaplan-Meier) à 30 mois ; ISTH : International Society on Thrombosis and Haemostasis ; 1x/j. : une fois par jour
Figure 2 : Délai avant première survenue du critère principal d’efficacité (accident vasculaire cérébral, infarctus du myocarde, décès cardiovasculaire) dans l’étude COMPASS
2x/j : deux fois par jour ; 1x/j : une fois par jour ; IC : intervalle de confiance
MC avec insuffisance cardiaque
Patients ayant récemment bénéficié d’une procédure de revascularisation du membre inférieur suite à une MAP symptomatique
Lors de l’étude pivot en double aveugle de phase III VOYAGER PAD, 6 564 patients ayant récemment bénéficié d’une procédure de revascularisation réussie (chirurgicale ou endovasculaire, procédures hybrides incluses) d’un membre inférieur suite à une MAP symptomatique ont été randomisés selon un rapport de 1/1 dans l’un des deux groupes de traitement antithrombotique : rivaroxaban 2,5 mg deux fois par jour en association avec l’AAS 100 mg une fois par jour, ou AAS 100 mg une fois par jour. Les patients étaient autorisés à recevoir en complément une dose standard de clopidogrel une fois par jour pendant 6 mois au maximum. L’objectif de l’étude était de démontrer l’efficacité et la sécurité du rivaroxaban en association avec l’AAS pour la prévention de l’infarctus du myocarde, des accidents vasculaire cérébraux ischémiques, des décès d’origine CV, de l’ischémie aiguë d’un membre ou des amputations majeures d’étiologie vasculaire chez les patients ayant récemment bénéficié d’une procédure de revascularisation réussie d’un membre inférieur suite à une MAP symptomatique. Les patients âgés de ≥ 50 ans présentant une MAP athéroscléreuse symptomatique modérée à sévère bien établie d’un membre inférieur , avec signes cliniques (à savoir, limitations fonctionnelles), anatomiques (à savoir, signes de MAP en aval de l’artère iliaque externe visibles à l’imagerie) et hémodynamiques (indice de pression systolique cheville-bras [ABI] ≤ 0,80 ou indice de pression systolique orteil-bras [TBI] ≤ 0,60 pour les patients sans antécédents de revascularisation d’un membre, ou ABI ≤ 0,85 ou TBI ≤ 0,65 pour les patients ayant des antécédents de revascularisation d’un membre) ont été inclus. Les patients nécessitant une bithérapie antiplaquettaire > 6 mois, tout traitement antiplaquettaire supplémentaire autre que l’AAS et le clopidogrel ou un traitement par anticoagulant oral, ainsi que les patients présentant des antécédents d’hémorragie intracrânienne, d’AVC ou d’AIT, et les patients présentant un DFGe < 15 mL/min ont été exclus.
La durée moyenne de suivi était de 24 mois et la durée maximale, de 4,1 ans. L’âge moyen des patients inclus était de 67 ans, et 17 % des patients de cette population étaient âgés de > 75 ans. Le temps médian entre la procédure de revascularisation de référence et le début du traitement à l’étude était de 5 jours dans la population globale (6 jours après une procédure de revascularisation chirurgicale et 4 jours après une procédure de revascularisation endovasculaire, procédures hybrides incluses). Dans l’ensemble, 53,0 % des patients ont reçu un traitement de fond par clopidogrel de courte durée, avec une durée médiane de 31 jours. Conformément au protocole de l’étude, le traitement à l’étude pouvait être instauré dès que possible, mais au plus tard 10 jours après une procédure de revascularisation réussie remplissant les critères de l’étude et une fois que l’hémostase avait été obtenue.
Le traitement par rivaroxaban 2,5 mg deux fois par jour en association avec l’AAS 100 mg une fois par jour était supérieur en termes de réduction du critère composite principal incluant infarctus du myocarde, accidents vasculaire cérébraux ischémiques, décès d’origine CV, ischémies aiguës d’un membre ou amputations majeures d’étiologie vasculaire par rapport à l’AAS seul (voir Tableau 9). Le critère principal de sécurité, à savoir les événements hémorragiques majeurs selon la définition TIMI, a montré une augmentation chez les patients traités par rivaroxaban et AAS, sans augmentation des hémorragies fatales ou intracrâniennes (voir Tableau 10).
Les critères secondaires d’efficacité ont été testés par ordre hiérarchique prédéfini (voir Tableau 9).
Tableau 9 : Données d’efficacité de l’étude de phase III VOYAGER PAD
|
Population de l’étude |
Patients ayant récemment bénéficié d’une procédure de revascularisation du membre inférieur suite à une MAP symptomatiquea) |
||
|
Posologie |
Rivaroxaban 2,5 mg 2x/j en association avec de l’AAS 100 mg 1x/j N = 3 286 n (risque cum. %)c) |
AAS 100 mg 1x/j
N = 3 278 n (risque cum. %)c) |
Hazard Ratio (IC à 95 %)d) |
|
Critère d’efficacité principalb) |
508 (15,5 %) |
584 (17,8 %) |
0,85 (0,76 ; 0,96) p = 0,0043e)* |
|
IDM |
131 (4,0 %) |
148 (4,5 %) |
0,88 (0,70 ; 1,12) |
|
Accident vasculaire cérébral ischémique |
71 (2,2 %) |
82 (2,5 %) |
0,87 (0,63 ; 1,19) |
|
Décès d’origine CV |
199 (6,1 %) |
174 (5,3 %) |
1,14 (0,93 ; 1,40) |
|
Ischémie aiguë d’un membref) |
155 (4,7 %) |
227 (6,9 %) |
0,67 (0,55 ; 0,82) |
|
Amputation majeure d’étiologie vasculaire |
103 (3,1 %) |
115 (3,5 %) |
0,89 (0,68 ; 1,16) |
|
Critère secondaire d’efficacité |
|
|
|
|
Revascularisation du membre de référence non planifiée pour récidive de l’ischémie du membre |
584 (17,8 %) |
655 (20,0 %) |
0,88 (0,79 ; 0,99) p = 0,0140e)* |
|
Hospitalisation pour événement thrombotique d’origine coronarienne ou périphérique (dans l’un ou l’autre des membres inférieurs) |
262 (8,0 %) |
356 (10,9 %) |
0,72 (0,62 ; 0,85) p < 0,0001e)* |
|
Mortalité toutes causes |
321 (9,8 %) |
297 (9,1 %) |
1,08 (0,92 ; 1,27) |
|
ETEV |
25 (0,8 %) |
41 (1,3 %) |
0,61 (0,37 ; 1,00) |
a) Analyse sur la population en intention de traiter, analyses principales ; adjudiqués par le CACI.
b) Critère composite : IDM, accident vasculaire cérébral ischémique, décès d’origine CV (décès CV et décès de cause inconnue), IAM et amputation majeure d’étiologie vasculaire.
c) Seule la première survenue de l’événement analysé d’un patient est prise en compte.
d) Le HR (IC à 95 %) repose sur le modèle des risques proportionnels de Cox stratifié par type de procédure et par utilisation du clopidogrel avec le traitement en tant que seule covariable.
e) La valeur p unilatérale est basée sur un test logarythmique de stratification par type de procédure et par l’utilisation du clopidogrel avec le traitement.
f) L’ischémie aiguë d’un membre est définie comme une dégradation nette et brutale de la perfusion d’un membre, soit associée à un pouls déficitaire d’apparition récente, soit nécessitant une intervention thérapeutique (c.-à-d. thrombolyse ou thrombectomie, ou revascularisation d’urgence), et conduisant à une hospitalisation.
* La réduction du critère d’efficacité était statistiquement supérieure.
1x/j : une fois par jour ; 2x/j : deux fois par jour ; CACI : comité d’adjudication clinique indépendant ; CV : cardiovasculaire ; IAM : ischémie aiguë d’un membre ; IC : intervalle de confiance ; IDM : infarctus du myocarde
Tableau 10 : Données de sécurité de l’étude de phase III VOYAGER PAD
|
Population de l’étude |
Patients ayant récemment bénéficié d’une procédure de revascularisation du membre inférieur pour une MAP symptomatiquea) |
||
|
Posologie |
Rivaroxaban 2,5 mg 2x/j en association avec de l’AAS 100 mg 1x/j N = 3 256 n (risque cum. %)b) |
AAS 100 mg 1x/j
N = 3 248 n (risque cum. %)b) |
Hazard Ratio (IC à 95 %)c
valeur pd) |
|
Hémorragie majeure selon la définition TIMI (associée ou non à un pontage coronarien) |
62 (1,9 %) |
44 (1,4 %) |
1,43 (0,97 ; 2,10) p = 0,0695 |
|
- Hémorragie fatale |
6 (0,2 %) |
6 (0,2 %) |
1,02 (0,33 ; 3,15) |
|
- Hémorragie intracrânienne |
13 (0,4 %) |
17 (0,5 %) |
0,78 (0,38 ; 1,61) |
|
- Hémorragie manifeste associée à une baisse du taux Hb ≥ 5 g/dL / Hct ≥ 15 % |
46 (1,4 %) |
24 (0,7 %) |
1,94 (1,18 ; 3,17) |
|
Hémorragie majeure selon la classification de l’ISTH |
140 (4,3 %) |
100 (3,1 %) |
1,42 (1,10 ; 1,84) p = 0,0068 |
|
- Hémorragie fatale |
6 (0,2 %) |
8 (0,2 %) |
0,76 (0,26 ; 2,19) |
|
- Hémorragie d’organe critique non fatale |
29 (0,9 %) |
26 (0,8 %) |
1,14 (0,67 ; 1,93) |
|
Événements hémorragiques non majeurs cliniquement pertinents selon la classification de l’ISTH |
246 (7,6 %) |
139 (4,3 %) |
1,81 (1,47 ; 2,23) |
a) Population d’analyse de la sécurité (tous les patients randomisés ayant reçu au moins une dose du médicament de l’étude), CACI : comité d’adjudication clinique indépendant
b) n = nombre de patients ayant présenté des événements, N = nombre de patients à risque, % = 100 * n/N, n/100 p-années = ratio du nombre de patients ayant présenté de nouveaux événements / durée cumulée d’exposition au risque.
c) Le HR (IC à 95 %) repose sur le modèle des risques proportionnels de Cox stratifié par type de procédure et par utilisation du clopidogrel avec le traitement en tant que seule covariable.
d) La valeur p bilatérale est basée sur un test logarithmique de stratification par type de procédure et par l’utilisation du clopidogrel avec le traitement.
MC avec insuffisance cardiaque
L’étude COMMANDER HF a inclus 5 022 patients présentant une insuffisance cardiaque ainsi qu’une maladie coronarienne (MC) significative à la suite d’une hospitalisation pour insuffisance cardiaque (IC) décompensée, répartis aléatoirement dans l’un des deux groupes de traitement : rivaroxaban 2,5 mg deux fois par jour (N = 2 507) ou placebo (N = 2 515). La durée médiane globale du traitement à l'étude a été de 504 jours.
Les patients devaient avoir présenté une IC symptomatique depuis au moins 3 mois et une fraction d'éjection ventriculaire gauche (FEVG) de ≤ 40 % dans l'année suivant le recrutement. A l’inclusion, la fraction médiane d'éjection était de 34 % (IQR : 28 %– 38 %) et 53 % des sujets étaient de classe NYHA III ou IV.
L'analyse principale d'efficacité (c.-à-d. le critère composite de la mortalité toutes causes confondues, de l'IDM ou de l'accident vasculaire cérébral) n'a révélé aucune différence statistiquement significative entre le groupe rivaroxaban 2,5 mg 2x/j et le groupe placebo avec un HR = 0,94 (IC à 95 % : 0,84– 1,05), p = 0,270. Concernant la mortalité toutes causes confondues, il n'y a pas eu de différence entre le rivaroxaban et le placebo quant au nombre d'événements (taux d'événements pour 100 patients-années ; 11,41 vs 11,63, HR : 0,98 ; IC à 95 % : 0,87 à 1,10 ; p = 0,743). Les taux d'événements pour l’IDM pour 100 patients-années (rivaroxaban vs placebo) ont été de 2,08 vs 2,52 (HR 0,83 ; IC 95 % : 0,63 à 1,08 ; p = 0,165) et pour l'accident vasculaire cérébral les taux d'événements pour 100 patients-années étaient de 1,08 vs 1,62 (HR : 0,66 ; IC à 95 % : 0,47 à 0,95 ; p = 0,023). Le critère principal de tolérance (c.-à-d. l'ensemble des saignements fatals ou des saignements à localisation critique avec un risque d'incapacité permanente) a été observé chez respectivement 18 (0,7 %) patients du groupe rivaroxaban 2,5 mg deux fois par jour et chez 23 (0,9 %) patients du groupe placebo (HR = 0,80 ; IC à 95 % : 0,43– 1,49 ; p = 0,484). Une augmentation statistiquement significative des saignements majeurs selon la classification de l'ISTH a été observée dans le groupe rivaroxaban comparativement au placebo (taux d'événements pour 100 patients-années : 2,04 vs 1,21, HR 1,68 ; IC à 95 % : 1,18 à 2,39 ; p = 0,003).
Chez les patients atteints d'insuffisance cardiaque légère ou modérée, les effets du traitement pour le sous-groupe COMPASS étaient semblables à ceux de l'ensemble de la population de l'étude (voir la section MC/MAP).
Patients à haut risque d’événements thromboemboliques présentant un syndrome des antiphospholipides positif aux trois tests
Dans le cadre d’une étude sponsorisée, menée par un investigateur indépendant, multicentrique, randomisée en ouvert avec adjudication en aveugle des critères d’évaluation, le rivaroxaban a été comparé à la warfarine chez des patients présentant des antécédents de thrombose auxquels a été diagnostiqué un syndrome des antiphospholipides et présentant un risque élevé d’événements thromboemboliques (patients triple positifs à l’ensemble des 3 tests antiphospholipides : anticoagulant circulant lupique, anticorps anticardiolipine et anticorps anti-bêta 2-glycoprotéine I). L’essai a été arrêté prématurément après la participation de 120 patients en raison d’un nombre excessif d’événements thromboemboliques survenus chez les patients du groupe rivaroxaban. Le suivi moyen était de 569 jours. 59 patients ont été randomisés pour recevoir du rivaroxaban 20 mg (15 mg pour les patients avec une clairance de la créatinine [ClCr] < 50 mL/min) et 61, de la warfarine (INR 2,0– 3,0). Des événements thromboemboliques sont survenus chez 12 % des patients randomisés sous rivaroxaban (4 accidents ischémiques cérébraux et 3 infarctus du myocarde). Aucun événement n’a été signalé chez les patients randomisés sous warfarine. 4 patients (7 %) du groupe rivaroxaban et 2 patients (3 %) du groupe warfarine ont présenté des saignements majeurs.
Population pédiatrique
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec du rivaroxaban dans tous les sous-groupes de la population pédiatrique dans la prévention des événements thromboemboliques (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
Le rivaroxaban est rapidement absorbé et les concentrations maximales (Cmax) sont obtenues 2 à 4 heures après la prise du comprimé.
L’absorption orale du rivaroxaban est presque totale et la biodisponibilité orale est élevée (80 à 100 %) pour le comprimé de 2,5 mg et le comprimé de 10 mg, qu’il soit pris au cours ou en dehors des repas. L’absorption d’aliments n’a pas d’effet sur l’ASC ou la Cmax du rivaroxaban pris à une dose de 2,5 mg et de 10 mg. Les comprimés de 2,5 mg et 10 mg de rivaroxaban peuvent être pris au cours ou en dehors des repas.
Les propriétés pharmacocinétiques du rivaroxaban sont pratiquement linéaires jusqu’à la dose de 15 mg environ par jour. À des doses plus élevées, l’absorption du rivaroxaban est limitée par sa dissolution, de ce fait, la biodisponibilité du rivaroxaban ainsi que son taux d’absorption diminuent avec l’augmentation de la dose. Ce phénomène est plus marqué lors d’une prise à jeun que lors d’une prise avec des aliments. La variabilité des caractéristiques pharmacocinétiques du rivaroxaban est modérée, avec une variabilité interindividuelle (CV%) comprise entre 30 % et 40 %.
L’absorption du rivaroxaban dépend de son site de libération dans le tractus gastro-intestinal. Par comparaison avec le comprimé, une diminution de 29 % et 56 % de l’ASC et de la Cmax a été observée lorsque le rivaroxaban sous forme de granulés est libéré dans la portion proximale de l’intestin grêle. L’exposition est davantage réduite lorsque le rivaroxaban est libéré dans la portion distale de l’intestin grêle ou dans le côlon ascendant. Par conséquent, l’administration du rivaroxaban en aval de l’estomac doit être évitée car elle peut entraîner une réduction de l’absorption du rivaroxaban et de l’exposition associée.
La biodisponibilité (ASC et Cmax) d’un comprimé entier de 20 mg de rivaroxaban et la biodisponibilité d’un comprimé écrasé, de même dosage, mélangé à de la compote de pommes et administré par voie orale ou mis en suspension dans de l’eau, puis administré au moyen d’une sonde gastrique et suivi d’un repas liquide, sont comparables. Étant donné que le profil pharmacocinétique du rivaroxaban est prévisible et proportionnel à la dose, les données de biodisponibilité issues de cette étude peuvent probablement être extrapolées aux doses inférieures de rivaroxaban.
Distribution
Le niveau de liaison avec les protéines plasmatiques chez l’Homme est élevé, environ 92 % à 95 %, la liaison se faisant essentiellement avec l’albumine sérique. Le volume de distribution est modéré : le Véq. est d’environ 50 litres.
Biotransformation et élimination
Sur l’ensemble de la dose de rivaroxaban administrée, 2/3 environ subissent une dégradation par voie métabolique, la moitié étant ensuite éliminée par voie rénale et l’autre moitié par voie fécale. Le tiers restant de la dose administrée subit une excrétion rénale directe dans les urines sous forme inchangée, essentiellement par sécrétion rénale active.
La métabolisation du rivaroxaban se déroule via le CYP3A4, le CYP2J2 et des mécanismes indépendants des CYP. La dégradation par oxydation de la fraction morpholinone et l’hydrolyse des liaisons amides sont les principaux points de biotransformation. D’après les études in vitro, le rivaroxaban est un substrat des protéines de transport P-gp (glycoprotéine-P) et BCRP (breast cancer resistance protein, protéine de résistance au cancer du sein).
Le rivaroxaban sous forme inchangée est le principal composant retrouvé dans le plasma humain, aucun métabolite majeur ou actif n’étant présent dans la circulation. Sa clairance systémique étant d’environ 10 L/h, le rivaroxaban peut être classé comme une substance à faible clairance. Après administration par voie intraveineuse d’une dose de 1 mg, la demi-vie d’élimination est d’environ 4,5 heures. Après administration par voie orale, l’élimination est limitée par le taux d’absorption. L’élimination du rivaroxaban hors du plasma se fait avec une demi-vie terminale de 5 à 9 heures chez les personnes jeunes et avec une demi-vie terminale de 11 à 13 heures chez les personnes âgées.
Populations particulières
Sexe
Aucune différence cliniquement pertinente n’a été notée entre les hommes et les femmes quant aux caractéristiques pharmacocinétiques et pharmacodynamiques.
Personnes âgées
Des concentrations plasmatiques plus élevées ont été observées chez les patients âgés par rapport à des patients plus jeunes, avec une ASC moyenne environ 1,5 fois supérieure, principalement en raison de la réduction de la clairance totale (apparente) et rénale. Aucun ajustement posologique n’est nécessaire.
Poids
Les poids extrêmes (< 50 kg ou > 120 kg) n’ont eu qu’une incidence mineure sur les concentrations plasmatiques en rivaroxaban (moins de 25 %). Aucun ajustement posologique n’est nécessaire.
Différences inter-ethniques
Aucune différence inter-ethnique cliniquement pertinente n’a été relevée entre les populations caucasiennes, afro-américaines, hispaniques, japonaises ou chinoises quant aux caractéristiques pharmacocinétiques et pharmacodynamiques du rivaroxaban.
Insuffisance hépatique
Chez les patients cirrhotiques atteints d’insuffisance hépatique légère (classe A de Child Pugh), les modifications des caractéristiques pharmacocinétiques du rivaroxaban observées étaient mineures (multiplication par 1,2 en moyenne de l’ASC du rivaroxaban), d’amplitude comparable à celles observées chez les sujets sains du groupe témoin. Chez les patients cirrhotiques atteints d’insuffisance hépatique modérée (classe B de Child Pugh), l’ASC moyenne du rivaroxaban a été multipliée par 2,3, augmentation significative par rapport aux volontaires sains. L’ASC du produit non lié a été multipliée par 2,6. Ces patients ont également présenté une élimination réduite du rivaroxaban par voie rénale, tout comme les patients atteints d’une insuffisance rénale modérée. Aucune donnée n’est disponible concernant les patients atteints d’insuffisance hépatique sévère.
L'inhibition de l’activité du facteur Xa a été augmentée d’un facteur 2,6 chez les patients atteints d’insuffisance hépatique modérée par rapport aux volontaires sains ; l’allongement du TQ a connu une augmentation similaire (multiplié par 2,1). Les patients atteints d’insuffisance hépatique modérée ont été plus sensibles au rivaroxaban, avec pour conséquence une pente du rapport PK/PD plus marquée entre la concentration et le TQ.
Le rivaroxaban est contre-indiqué chez les patients présentant une atteinte hépatique associée à une coagulopathie et à un risque de saignement cliniquement significatif, y compris chez les patients cirrhotiques avec un score de Child Pugh classe B ou C (voir rubrique 4.3).
Insuffisance rénale
Il a été observé un lien entre l’augmentation de l’exposition au rivaroxaban et la diminution de la fonction rénale évaluée par la mesure de la clairance de la créatinine (ClCr). En cas d’insuffisance rénale légère (ClCr de 50 à 80 mL/min), modérée (ClCr de 30 à 49 mL/min) ou sévère (ClCr de 15 à 29 mL/min), les concentrations plasmatiques du rivaroxaban (ASC) ont été multipliées respectivement par 1,4 ; 1,5 et 1,6. Les augmentations correspondantes des effets pharmacodynamiques ont été plus marquées. En cas d’insuffisance rénale légère, modérée ou sévère, l'inhibition globale de l’activité du facteur Xa a été augmentée respectivement d’un facteur 1,5 ; 1,9 et 2,0 par rapport aux volontaires sains ; l’allongement du TQ a connu une augmentation similaire, multiplié respectivement par 1,3 ; 2,2 et 2,4. Aucune donnée n’est disponible concernant les patients dont la clairance de la créatinine est < 15 mL/min.
Étant donné la forte liaison du rivaroxaban aux protéines plasmatiques, le produit n’est probablement pas dialysable.
L’utilisation du produit n’est pas recommandée chez les patients avec une clairance de la créatinine < 15 mL/min. Le rivaroxaban doit être utilisé avec prudence chez les patients dont la clairance de la créatinine est de 15 à 29 mL/min (voir rubrique 4.4).
Données pharmacocinétiques chez les patients
Chez les patients ayant reçu du rivaroxaban à la dose de 2,5 mg deux fois par jour pour la prévention des événements athérothrombotiques suite à un SCA, la concentration moyenne géométrique (intervalle prédictif de 90 %) 2 à 4 h et environ 12 h après la dose (représentant approximativement les concentrations maximales et minimales durant l’intervalle entre les doses) était respectivement de 47 mcg/L (13– 123) et 9,2 mcg/L (4,4– 18).
Relations pharmacocinétique/pharmacodynamique
Le rapport pharmacocinétique/pharmacodynamique (PK/PD) entre la concentration plasmatique du rivaroxaban et plusieurs critères d’évaluation PD (inhibition du facteur Xa, TQ, TCA, Heptest) a été évalué après administration d’une large gamme de doses (de 5 à 30 mg deux fois par jour). Le rapport entre la concentration de rivaroxaban et l’activité du facteur Xa a été très bien décrit par un modèle Emax. En ce qui concerne le TQ, le modèle linéaire fournit généralement une meilleure description des données. La pente varie considérablement en fonction des thromboplastines utilisées.
Par exemple, lorsque le réactif Néoplastine est utilisé, le TQ de référence est d’environ 13 s et la pente est d’environ 3 à 4 s/(100 mcg/L). Les résultats des analyses PK/PD en phase II et III sont cohérents avec les données établies chez les sujets sains.
Population pédiatrique
La sécurité et l’efficacité du médicament n’ont pas été établies dans les indications de SCA et de MC/MAP chez les enfants et adolescents âgés de 0 à 18 ans.
5.3. Données de sécurité préclinique
Les effets observés au cours des études de toxicité en administration répétée étaient principalement dus à l’exacerbation de l’activité pharmacodynamique du rivaroxaban. Chez le rat, une augmentation des taux plasmatiques d’IgG et d’IgA a été observée à des niveaux d’exposition cliniquement pertinents.
Chez le rat, aucun effet sur la fertilité des mâles ou des femelles n’a été observé. Les études chez l’animal ont montré une toxicité sur la reproduction liée au mode d’action pharmacologique du rivaroxaban (complications hémorragiques, par ex.). Une toxicité embryo-fœtale (fausse-couche post-implantatoire, retard/progression de l’ossification, taches hépatiques multiples de couleur claire) et une incidence accrue des malformations courantes ainsi que des modifications placentaires ont été observées à des taux plasmatiques cliniquement pertinents. Lors de l’étude prénatale et postnatale chez le rat, une réduction de la viabilité de la descendance a été observée à des doses toxiques pour la mère.
Lactose monohydraté, cellulose microcristalline, croscarmellose sodique, hypromellose, laurilsulfate de sodium, stéarate de magnésium.
Pelliculage du comprimé
Hypromellose, dioxyde de titane (E171), macrogol, oxyde de fer jaune (E172).
2 ans
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Les comprimés pelliculés sont présentés sous plaquettes (PVC/Aluminium).
Taille du conditionnement : 20, 28, 56, 100, 168 et 196 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Ecrasement de comprimés
Les comprimés de rivaroxaban peuvent être écrasés et mis en suspension dans 50 mL d’eau, puis être administrés au moyen d’une sonde naso-gastrique ou d’une sonde de gastrostomie après confirmation du bon positionnement gastrique de la sonde. La sonde doit ensuite être rincée avec de l’eau. Puisque l’absorption du rivaroxaban dépend du site de libération du médicament, il convient d’éviter l’administration du rivaroxaban en aval de l’estomac, car cela peut entraîner une absorption réduite et, de ce fait, une exposition réduite au médicament. Une alimentation entérale n’est pas nécessaire immédiatement après l’administration des comprimés de 2,5 mg.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
35 RUE DU VAL DE MARNE
75013 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 984 2 1 : 20 comprimés sous plaquettes (PVC/Aluminium).
· 34009 301 984 3 8 : 28 comprimés sous plaquettes (PVC/Aluminium).
· 34009 301 984 4 5 : 56 comprimés sous plaquettes (PVC/Aluminium).
· 34009 550 706 9 6 : 100 comprimés sous plaquettes (PVC/Aluminium).
· 34009 550 707 0 2 : 168 comprimés sous plaquettes (PVC/Aluminium).
· 34009 550 707 1 9 : 196 comprimés sous plaquettes (PVC/Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 21/03/2024
RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
Rivaroxaban
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé ?
3. Comment prendre RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
RIVAROXABAN ZENTIVA vous a été prescrit parce que
· un syndrome coronarien aigu vous a été diagnostiqué (un ensemble de maladies incluant crise cardiaque et angor instable, correspondant à un type de douleur thoracique sévère) et parce que votre prise de sang a montré une augmentation des enzymes cardiaques.
Chez l’adulte, RIVAROXABAN ZENTIVA réduit le risque de présenter à nouveau une crise cardiaque ou de décéder des suites d’une maladie liée au cœur ou aux vaisseaux sanguins.
RIVAROXABAN ZENTIVA vous sera prescrit avec un autre médicament. Ainsi, votre médecin vous demandera également de prendre :
o de l’acide acétylsalicylique ou
o de l’acide acétylsalicylique plus du clopidogrel ou de la ticlopidine.
ou
· un risque élevé de présenter un caillot sanguin en raison d’une maladie coronarienne ou d’une maladie artérielle périphérique qui provoque des symptômes vous a été diagnostiqué.
Chez l’adulte, RIVAROXABAN ZENTIVA réduit le risque de présenter des caillots sanguins (événements athérothrombotiques).
RIVAROXABAN ZENTIVA vous sera prescrit avec un autre médicament. Ainsi, votre médecin vous demandera également de prendre de l’acide acétylsalicylique.
Dans certains cas, si vous recevez RIVAROXABAN ZENTIVA après une procédure consistant à désobstruer une artère rétrécie ou obstruée dans votre jambe pour restaurer la circulation sanguine, votre médecin pourra également vous prescrire du clopidogrel à prendre en plus de l’acide acétylsalicylique pendant une courte durée.
RIVAROXABAN ZENTIVA contient une substance active appelée rivaroxaban et appartient à une classe de médicaments appelés antithrombotiques. Il agit en bloquant un facteur de la coagulation sanguine (le facteur Xa), réduisant ainsi la tendance du sang à former des caillots.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé ?
Ne prenez jamais RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
· si vous êtes allergique au rivaroxaban ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· si vous êtes sujet à des saignements excessifs ;
· si vous avez une maladie ou une prédisposition au niveau d’un organe qui augmente les risques de saignements importants (par exemple, un ulcère de l’estomac, une blessure ou un saignement dans le cerveau, une intervention chirurgicale récente du cerveau ou des yeux) ;
· si vous prenez déjà des médicaments pour empêcher la formation de caillots sanguins (warfarine, dabigatran, apixaban ou héparine, par ex.), sauf lorsque vous changez de traitement anticoagulant ou si vous avez une voie d’abord artérielle ou veineuse par laquelle de l’héparine vous est administrée pour la maintenir ouverte ;
· si vous avez un syndrome coronarien aigu et que vous avez déjà eu un saignement ou un caillot sanguin dans le cerveau (accident vasculaire cérébral, AVC) ;
· si vous avez une maladie coronarienne ou une maladie artérielle périphérique et que vous avez déjà présenté un saignement au niveau du cerveau (accident vasculaire cérébral) ou un blocage des petites artères amenant le sang aux tissus profonds du cerveau (AVC lacunaire) ou un caillot de sang dans le cerveau (AVC lacunaire ischémique, non lacunaire) au cours du mois précédent ;
· si vous présentez une maladie du foie augmentant les risques de saignement ;
· si vous êtes enceinte ou si vous allaitez.
Si vous êtes dans l’une de ces situations, ne prenez pas RIVAROXABAN ZENTIVA et prévenez votre médecin.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre RIVAROXABAN ZENTIVA.
A part l’acide acétylsalicylique et le clopidogrel ou la ticlopidine, RIVAROXABAN ZENTIVA ne doit pas être utilisé en association avec d’autres médicaments utilisés pour fluidifier le sang, tels que le prasugrel ou le ticagrelor.
Faites attention avec RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
· si vous présentez un risque accru de saignement, ce qui pourrait être le cas dans les situations suivantes :
o insuffisance rénale sévère, car l’état de votre fonction rénale peut avoir une influence sur la quantité de médicament pouvant agir dans votre corps,
o si vous prenez d’autres médicaments destinés à prévenir la formation de caillots sanguins (warfarine, dabigatran, apixaban ou héparine, par ex.), lorsque vous changez de traitement anticoagulant ou si vous avez une voie d’abord artérielle ou veineuse par laquelle de l’héparine vous est administrée pour la maintenir ouverte (voir rubrique « Autres médicaments et RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé »),
o troubles hémorragiques,
o pression artérielle très élevée, non contrôlée par un traitement médicamenteux,
o maladie de l’estomac ou de l’intestin susceptible d’entraîner un saignement, par ex. inflammation des intestins ou de l’estomac, ou inflammation de l’œsophage par ex. due à un reflux gastro-œsophagien (remontées acides de l’estomac vers l’œsophage) ou des tumeurs localisées dans l'estomac, les intestins, l'appareil génital ou l'appareil urinaire,
o anomalies au niveau des vaisseaux sanguins du fond de l’œil (rétinopathie),
o maladie des poumons avec dilatation des bronches et présence de pus (bronchectasie) ou antécédents de saignement dans les poumons,
o âge supérieur à 75 ans,
o poids inférieur à 60 kg,
o si vous présentez une maladie coronarienne accompagnée d’une insuffisance cardiaque sévère symptomatique ;
· si vous avez une valve cardiaque artificielle ;
· si vous savez que vous présentez une maladie appelée syndrome des antiphospholipides (une affection du système immunitaire qui cause un risque accru de formation de caillots sanguins), dites-le à votre médecin; il décidera si le traitement doit éventuellement être modifié.
Si vous êtes dans l’une de ces situations, prévenez votre médecin avant de prendre RIVAROXABAN ZENTIVA. Votre médecin décidera si vous devez être traité(e) par ce médicament et s’il est nécessaire de vous surveiller étroitement.
Si vous devez bénéficier d’une intervention chirurgicale
· il est très important que vous preniez RIVAROXABAN ZENTIVA exactement à l’heure indiquée par votre médecin avant et après l’opération ;
· si l’intervention chirurgicale implique l’utilisation d’un cathéter ou une injection dans votre colonne vertébrale (pour une anesthésie péridurale ou rachidienne ou pour l’injection d’un calmant, par ex.) :
o il est très important que vous preniez RIVAROXABAN ZENTIVA exactement à l’heure indiquée par votre médecin avant et après l’injection ou le retrait du cathéter ;
o prévenez immédiatement votre médecin si vous ressentez un engourdissement ou une faiblesse dans les jambes ou des problèmes au niveau des intestins ou de la vessie après la fin de l’anesthésie car des soins urgents sont nécessaires.
Enfants et adolescents
L’utilisation de comprimés pelliculés de RIVAROXABAN ZENTIVA 2,5 mg n’est pas recommandée chez les patients âgés de moins de 18 ans. Les informations disponibles concernant leur utilisation chez l’enfant et l’adolescent ne sont pas suffisantes.
Autres médicaments et RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris un médicament obtenu sans ordonnance.
· Si vous prenez :
o certains médicaments contre les infections fongiques ou mycoses (le fluconazole, l’itraconazole, le voriconazole, le posaconazole, par ex.), sauf s’ils sont uniquement appliqués sur la peau ;
o des comprimés de kétoconazole (utilisés pour traiter le syndrome de Cushing – lorsque le corps produit une quantité trop importante de cortisol) ;
o certains médicaments contre les infections bactériennes (clarithromycine, érythromycine, par ex.) ;
o certains antiviraux contre le VIH/SIDA (le ritonavir, par ex.) ;
o d’autres médicaments utilisés pour fluidifier le sang (par ex., l’énoxaparine, le clopidogrel ou les anti-vitamine K tels que la warfarine et l’acénocoumarol, le prasugrel et le ticagrelor [voir « Avertissements et précautions »]) ;
o des anti-inflammatoires et des anti-douleurs (le naproxène ou l’acide acétylsalicylique, par ex.) ;
o de la dronédarone, un médicament utilisé pour traiter les troubles du rythme cardiaque ;
o certains médicaments utilisés pour traiter la dépression (inhibiteurs sélectifs de la recapture de la sérotonine [ISRS] ou inhibiteurs de la recapture de la sérotonine et de la noradrénaline [IRSN]).
Si vous êtes dans l’une de ces situations, prévenez votre médecin avant de prendre RIVAROXABAN ZENTIVA car l’effet de RIVAROXABAN ZENTIVA pourrait être augmenté. Votre médecin décidera si vous devez être traité(e) par RIVAROXABAN ZENTIVA et si une surveillance étroite est nécessaire.
Si votre médecin pense que vous présentez un risque élevé de développer un ulcère de l’estomac ou de l’intestin, il peut également vous prescrire un traitement préventif contre les ulcères.
· Si vous prenez :
o certains médicaments utilisés pour traiter l'épilepsie (phénytoïne, carbamazépine, phénobarbital) ;
o du millepertuis (Hypericum perforatum), un produit à base de plante utilisé pour la dépression ;
o de la rifampicine, un antibiotique.
Si vous êtes dans l’une de ces situations, prévenez votre médecin avant de prendre RIVAROXABAN ZENTIVA car l’effet de RIVAROXABAN ZENTIVA pourrait être réduit. Votre médecin décidera si vous devez être traité(e) par RIVAROXABAN ZENTIVA et si une surveillance étroite est nécessaire.
RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé avec des aliments et boissons
Sans objet.
Ne prenez pas RIVAROXABAN ZENTIVA si vous êtes enceinte ou si vous allaitez. Si une grossesse est possible, utilisez un contraceptif fiable pendant la période où vous prenez RIVAROXABAN ZENTIVA. Si vous devenez enceinte pendant que vous prenez ce médicament, informez-en immédiatement votre médecin qui décidera de la conduite à tenir.
Conduite de véhicules et utilisation de machines
RIVAROXABAN ZENTIVA peut être à l’origine de sensations vertigineuses (effet indésirable fréquent) ou d’évanouissements (effet indésirable peu fréquent) (voir rubrique 4 « Quels sont les effets indésirables éventuels ? »). Ne conduisez pas, ne roulez pas à vélo ou n’utilisez pas d’outils ou de machines si vous êtes sujet à ces symptômes.
RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé contient du lactose monohydraté (un type de sucre) et du sodium
Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé ?
Quelle quantité prendre
La dose recommandée est d’un comprimé de 2,5 mg deux fois par jour. Prenez RIVAROXABAN ZENTIVA à peu près à heure fixe chaque jour (par exemple, un comprimé le matin et un le soir).
Ce médicament peut être pris au cours ou en dehors des repas.
Si vous avez des difficultés à avaler le comprimé entier, discutez avec votre médecin des autres moyens de prendre RIVAROXABAN ZENTIVA. Le comprimé peut être écrasé et mélangé à de l’eau ou de la compote de pommes, immédiatement avant la prise.
Si nécessaire, votre médecin pourra aussi vous donner le comprimé de RIVAROXABAN ZENTIVA écrasé en utilisant une sonde gastrique.
RIVAROXABAN ZENTIVA vous sera prescrit avec un autre médicament.
Ainsi, votre médecin vous demandera également de prendre de l’acide acétylsalicylique. Si l’on vous prescrit RIVAROXABAN ZENTIVA après un syndrome coronarien aigu, votre médecin vous demandera également de prendre du clopidogrel ou de la ticlopidine.
Si vous recevez RIVAROXABAN ZENTIVA après une procédure consistant à désobstruer une artère rétrécie ou obstruée dans votre jambe pour restaurer la circulation sanguine, votre médecin pourra également vous prescrire du clopidogrel à prendre en plus de l’acide acétylsalicylique pendant une courte durée.
Votre médecin vous indiquera quelle dose de ces médicaments vous devez prendre (habituellement entre 75 et 100 mg d’acide acétylsalicylique par jour ou une dose quotidienne de 75 à 100 mg d’acide acétylsalicylique plus une dose quotidienne de 75 mg de clopidogrel ou une dose quotidienne standard de ticlopidine).
A quel moment commencer RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
Le traitement par RIVAROXABAN ZENTIVA après un syndrome coronarien aigu devra être débuté dès que possible après la stabilisation du syndrome coronarien aigu, au plus tôt 24 heures après l’admission à l’hôpital et au moment où le traitement anticoagulant parentéral (via injection) aurait normalement été arrêté.
Votre médecin vous dira quand commencer votre traitement par RIVAROXABAN ZENTIVA si l’on vous a diagnostiqué une maladie coronarienne ou une maladie artérielle périphérique.
Votre médecin décidera de la durée de votre traitement.
Si vous avez pris plus de RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé que vous n’auriez dû
Contactez immédiatement votre médecin si vous avez pris trop de comprimés de RIVAROXABAN ZENTIVA. L’ingestion de trop de RIVAROXABAN ZENTIVA augmente le risque de saignement.
Si vous oubliez de prendre RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre. Si vous avez oublié une dose, prenez la dose suivante à l’heure habituelle.
Si vous arrêtez de prendre RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
Prenez RIVAROXABAN ZENTIVA avec régularité et aussi longtemps que votre médecin vous le prescrit.
N’arrêtez pas de prendre RIVAROXABAN ZENTIVA sans en avoir d’abord parlé avec votre médecin. Si vous arrêtez de prendre ce médicament, vous pourriez être exposé(e) à un plus fort risque de nouvelle crise cardiaque ou d’accident vasculaire cérébral ou de décès des suites d’une maladie liée à votre cœur ou à vos vaisseaux sanguins.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme les autres médicaments du même type réduisant la formation de caillots sanguins, RIVAROXABAN ZENTIVA peut provoquer des saignements pouvant éventuellement mettre la vie du patient en danger. Un saignement excessif peut entraîner une chute soudaine de la pression artérielle (choc). Dans certains cas, ces saignements peuvent ne pas être apparents.
Prévenez immédiatement votre médecin si vous présentez l’un des effets indésirables suivants :
· Signes de saignement
o saignement dans le cerveau ou à l’intérieur du crâne (les symptômes peuvent inclure des maux de tête, une faiblesse d’un côté du corps, des vomissements, des convulsions, une diminution du niveau de conscience et une raideur de la nuque. Il s’agit d’une urgence médicale grave. Consultez immédiatement un médecin !)
o saignement abondant ou prolongé,
o sensations anormales de faiblesse, fatigue, pâleur, sensations vertigineuses, mal de tête, gonflement inexpliqué, essoufflement, douleur dans la poitrine ou angine de poitrine
Votre médecin pourra décider de surveiller plus étroitement votre santé ou de modifier votre traitement.
· Signes de réactions cutanées sévères
o une éruption cutanée intense et diffuse, des cloques ou des lésions des muqueuses, par exemple : dans la bouche ou les yeux (syndrome de Stevens-Johnson / nécrolyse épidermique toxique)
o une réaction médicamenteuse provoquant éruption cutanée, fièvre, inflammation des organes internes, anomalies du sang et maladie systémique (syndrome DRESS).
La fréquence de ces effets indésirables est très rare (jusqu’à une personne sur 10 000).
· Signes de réactions allergiques sévères
o gonflement du visage, des lèvres, de la bouche, de la langue ou de la gorge; difficulté à avaler; urticaire et difficultés respiratoires; chute brutale de la pression artérielle.
Les fréquences de ces réactions allergiques sévères sont très rares (réactions anaphylactiques, y compris choc anaphylactique; pouvant toucher jusqu’à une personne sur 10 000) et peu fréquentes (Œdème de Quincke et œdème allergique; pouvant toucher 1 personne sur 100).
Liste globale des effets indésirables éventuels
Fréquent (pouvant affecter jusqu’à 1 personne sur 10)
· diminution du nombre de globules rouges dans le sang pouvant entraîner une pâleur de la peau, une faiblesse ou un essoufflement ;
· saignement dans l’estomac ou les intestins, saignement urogénital (y compris sang dans les urines et menstruations [règles] plus abondantes), saignement de nez, saignement des gencives ;
· saignement dans les yeux (y compris saignement au niveau du blanc de l’œil) ;
· saignement des tissus ou saignement interne (hématome, bleus) ;
· toux avec expectoration (crachat) de sang ;
· saignement au niveau de la peau ou sous la peau ;
· saignement suite à une intervention chirurgicale ;
· suintement ou saignement au niveau de la plaie chirurgicale ;
· gonflement des membres ;
· douleur dans les membres ;
· altération du fonctionnement des reins (pouvant se voir sur les analyses demandées par votre médecin) ;
· fièvre ;
· douleur à l’estomac, indigestion, envie de vomir ou vomissements, constipation, diarrhée ;
· pression artérielle basse (les symptômes pouvant être des sensations vertigineuses ou un évanouissement lors du passage en position debout) ;
· diminution de la vivacité (fatigue, faiblesse), mal de tête, sensations vertigineuses ;
· éruption cutanée, démangeaisons ;
· possible élévation de certaines enzymes du foie lors des analyses de sang.
Peu fréquent (pouvant affecter jusqu’à 1 personne sur 100)
· saignement dans le cerveau ou à l’intérieur du crâne (voir plus haut, les signes de saignement);
· saignement au niveau d’une articulation entraînant douleur et gonflement ;
· thrombopénie (faible taux de plaquettes, qui sont les cellules qui permettent au sang de coaguler) ;
· réactions allergiques, y compris réactions allergiques au niveau de la peau ;
· altération du fonctionnement du foie (pouvant se voir sur les analyses demandées par votre médecin) ;
· possible élévation de la bilirubine, de certaines enzymes du pancréas ou du foie ou du nombre de plaquettes lors des analyses de sang ;
· évanouissement ;
· malaise général ;
· accélération des battements cardiaques ;
· bouche sèche ;
· urticaire.
Rare (pouvant affecter jusqu’à 1 personne sur 1 000)
· saignement dans un muscle ;
· cholestase (diminution de l’écoulement biliaire), hépatite dont lésion hépatocellulaire (inflammation du foie dont lésion du foie) ;
· coloration jaune de la peau et des yeux (jaunisse) ;
· gonflement localisé ;
· accumulation de sang (hématome) dans l’aine en complication d’une intervention cardiaque consistant à introduire un cathéter au niveau de l’artère de la jambe (pseudo-anévrisme).
Très rare (pouvant toucher jusqu’à 1 personne sur 10 000)
· accumulation d'éosinophiles, un type de globules blancs granulocytaires qui provoquent une inflammation des poumons (pneumonie à éosinophiles)
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
· insuffisance rénale suite à un saignement sévère ;
· augmentation de la pression dans les muscles des jambes ou des bras après un saignement, entraînant des douleurs, des gonflements, une modification de la sensibilité, un engourdissement ou une paralysie (syndrome de compression des loges faisant suite à un saignement).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la plaquette et la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé
· La substance active est :
Rivaroxaban.................................................................................................................... 2,5 mg
Pour un comprimé pelliculé.
· Les autres composants sont :
Noyau du comprimé : lactose monohydraté, cellulose microcristalline, croscarmellose sodique, hypromellose, laurilsulfate de sodium, stéarate de magnésium.
Pelliculage du comprimé : hypromellose, dioxyde de titane (E171), macrogol, oxyde de fer jaune (E172).
Qu’est-ce que RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé et contenu de l’emballage extérieur
RIVAROXABAN ZENTIVA 2,5 mg, comprimé pelliculé se présente sous forme de comprimés pelliculés jaunes, ronds, biconvexes (d’environ 5 mm de diamètre) comportant la mention « 2,5 » gravée en relief sur une face, l’autre face étant lisse.
RIVAROXABAN ZENTIVA 2,5 mg est disponible en boîtes de 20, 28, 56, 100, 168 et 196 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
35 RUE DU VAL DE MARNE
75013 PARIS
Exploitant de l’autorisation de mise sur le marché
35 RUE DU VAL DE MARNE
75013 PARIS
B-DUL THEODOR PALLADY NR. 50, SECTOR 3,
BUCAREST, COD 032266
ROUMANIE
ou
Pharmadox Healthcare Ltd.
KW20A Kordin Industrial Park
Paola, PLA3000
Malte
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants :
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
CARTE DE SURVEILLANCE DU PATIENT
Carte de Surveillance du Patient
Logo Zentiva France
RIVAROXABAN ZENTIVA 2,5 mg (cocher la case pour indiquer la dose prescrite)
RIVAROXABAN ZENTIVA 10 mg (cocher la case pour indiquer la dose prescrite)
RIVAROXABAN ZENTIVA 15 mg (cocher la case pour indiquer la dose prescrite)
RIVAROXABAN ZENTIVA 20 mg (cocher la case pour indiquer la dose prescrite)
Gardez toujours cette carte sur vous
Montrez cette carte à tout médecin ou dentiste que vous consultez avant de commencer un traitement
Je suis sous traitement anticoagulant par RIVAROXABAN ZENTIVA (rivaroxaban)
Nom :
Adresse :
Date de naissance :
Poids :
Autres médicaments/Pathologies :
En cas d’urgence, veuillez prévenir :
Nom du médecin :
Téléphone du médecin :
Cachet du médecin :
Veuillez aussi prévenir :
Nom :
Téléphone :
Lien avec le patient :
Information à destination des professionnels de santé :
· Les valeurs de l’INR ne doivent pas être utilisées car elles ne représentent pas une valeur fiable de la mesure de l’activité anticoagulante de RIVAROXABAN ZENTIVA.
Que faut-il savoir sur RIVAROXABAN ZENTIVA ?
· RIVAROXABAN ZENTIVA fluidifie le sang, ce qui vous protège de la formation de caillots sanguins dangereux pour la santé.
· RIVAROXABAN ZENTIVA doit être pris exactement comme votre médecin vous l’a prescrit. N’oubliez jamais de prendre votre traitement afin d’assurer une protection optimale contre les caillots sanguins.
· Vous ne devez jamais arrêter de prendre RIVAROXABAN ZENTIVA sans en parler d’abord à votre médecin, car le risque de formation de caillots sanguins peut être augmenté.
· Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, avant de prendre RIVAROXABAN ZENTIVA.
· Informez votre médecin que vous êtes traité par RIVAROXABAN ZENTIVA avant toute intervention chirurgicale ou tout geste invasif.
Quand dois-je demander conseil à mon médecin ou à mon pharmacien ?
Lors de la prise d’un médicament fluidifiant le sang comme RIVAROXABAN ZENTIVA, il est important de connaître les éventuels effets indésirables. Les saignements sont l’effet indésirable le plus fréquent. Si vous présentez un risque de saignement inhabituel, ne commencez pas à prendre RIVAROXABAN ZENTIVA sans en avoir parlé d’abord avec votre médecin. Prévenez immédiatement votre professionnel de santé si vous présentez des signes ou des symptômes de saignements tels que :
· douleurs
· gonflement ou sensations d’inconfort
· maux de tête, sensations vertigineuses ou faiblesse
· ecchymoses/bleus inhabituel(le)s, saignements du nez, saignements des gencives, ou saignement d’une plaie difficile à stopper
· menstruations (règles) ou saignements vaginaux plus abondants que d’ordinaire
· sang dans les urines pouvant les colorer en rose ou marron, selles rouges ou noires
· si vous crachez du sang, ou vomissez du sang ou des substances ressemblant à du marc de café
Comment dois-je prendre RIVAROXABAN ZENTIVA ?
· Pour garantir une protection optimale, RIVAROXABAN ZENTIVA
o 2,5 mg peut être pris au cours ou en dehors des repas,
o 10 mg peut être pris au cours ou en dehors des repas,
o 15 mg doit être pris au cours des repas,
o 20 mg doit être pris au cours des repas.