Dernière mise à jour le 01/06/2026
ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.)
Présentations
> 1 flacon(s) en verre de 1 g
Code CIP : 562 016-7 ou 34009 562 016 7 9
Déclaration de commercialisation : 29/01/2001
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
Autres informations
- Titulaire de l'autorisation : PFIZER HOLDING FRANCE
- Conditions de prescription et de délivrance :
- liste I
- prescription réservée aux spécialistes et services HEMATOLOGIE
- prescription réservée aux spécialistes et services MEDECINE INTERNE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- réservé à l'usage HOSPITALIER
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 037 500 1
ANSM - Mis à jour le : 14/04/2026
ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.)
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour un flacon de poudre.
Poudre pour solution pour perfusion (I.V.).
4.1. Indications thérapeutiques
· Leucémies aiguës myéloblastiques notamment en rechute.
· Leucémies aiguës myéloblastiques dites réfractaires (rechutant en cours de traitement).
· Leucémies aiguës lymphoblastiques en rechute, et leucémies secondaires.
4.2. Posologie et mode d'administration
La cytarabine doit être administrée en milieu hospitalier, sous stricte surveillance médicale.
Avant utilisation, la cytarabine peut être reconstituée avec le solvant ci-dessous :
· eau pour préparation injectable.
Les volumes de solvant à utiliser pour la reconstitution sont comme suit :
· cytarabine 500 mg est reconstituée avec 10 ml de solvant,
· cytarabine 1 g est reconstituée avec 10 ml de solvant,
· cytarabine 2 g est reconstituée avec 20 ml de solvant.
A ce fort dosage, la cytarabine 1 g est administrée en perfusion intraveineuse dans 250 ml de solution isotonique de glucose ou de solution isotonique de chlorure de sodium d'une durée de 1 à 3 heures, à une posologie de 2 à 3 g/m² toutes les 12 heures ; soit 4 à 6 g/m²/24 heures pendant 6 jours (soit 12 doses au total par cure).
Ne pas utiliser de solvant contenant de l’alcool benzylique.
ADAPTATION POSOLOGIQUE
· La fréquence des cures est fonction du résultat thérapeutique et de la toxicité hématologique et extra-hématologique.
· Des contrôles répétés, sanguins et médullaires devront être effectués, surtout en début de traitement. Les fonctions hépatiques et rénales seront également surveillées.
· L'adaptation de la posologie se fait en fonction des résultats des examens sanguins et médullaires (myélogramme).
· Habituellement, le traitement est interrompu si :
o les plaquettes sont inférieures à 50 000/mm3,
o les polynucléaires neutrophiles sont inférieurs à 1 000/mm3.
· La reprise du traitement se fait dès que les chiffres des numérations le permettent et dès que les cellules blastiques réapparaissent dans le sang ou dans la moelle. Le fait d'attendre la normalisation de la numération pour reprendre le traitement est préjudiciable au contrôle ultérieur de la maladie.
· Les posologies seront aussi modifiées en cas de phénomènes toxiques autres qu'hématologiques et en cas d'association à d'autres agents chimiothérapiques.
· La cytarabine peut être utilisée en monothérapie et en association. Différents schémas thérapeutiques ont été utilisés. L'ARA-C, à la dose de 3 g/m² en perfusion I.V. de 1 à 3 heures toutes les 12 heures pendant 4 à 6 jours, a pu être associée à de l'adriamycine (30 mg/m² J6 et J7), à de l'asparaginase (6 000 unités/m²), à de la rubidazone, à de l'AMSA (150 à 200 mg/m²/jour x 3), avec des résultats thérapeutiques significatifs. La toxicité hématologique est souvent plus prononcée, de même que la toxicité digestive, notamment sous forme de mucite.
Attention
Il est extrêmement important de s’assurer que l’administration est intraveineuse. Toute extravasation risquerait de produire une nécrose des tissus environnants. Dans ce cas, il convient d’interrompre immédiatement l’injection.
Modalités de manipulation
La préparation des solutions injectables de cytotoxiques doit être obligatoirement réalisée par un personnel spécialisé et entraîné ayant une connaissance des médicaments utilisés, dans des conditions assurant la protection de l’environnement et surtout la protection du personnel qui manipule. Elle nécessite un local de préparation réservé à cet usage. Il est interdit de fumer, de manger, de boire dans ce local. Les manipulateurs doivent disposer d’un ensemble de matériel approprié à la manipulation, notamment blouses à manches longues, masques de protection, calot, lunettes de protection, gants à usage unique stériles, champs de protection du plan de travail, conteneurs et sacs de collecte des déchets. Les excréta et les vomissures doivent être manipulés avec précaution. Les femmes enceintes doivent être averties et éviter la manipulation des cytotoxiques. Tout contenant cassé doit être traité avec les mêmes précautions et considéré comme un déchet contaminé. L’élimination des déchets contaminés se fait par incinération dans des conteneurs rigides étiquetés à cet effet.
Ces dispositions peuvent être envisagées dans le cadre du réseau de cancérologie (circulaire DGS/DH/98 n°98/188 du 24 mars 1998) en collaboration avec toute structure adaptée et remplissant les conditions requises.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Celles communes à toute thérapeutique cytotoxique.
· Aplasie médullaire préexistante.
· Encéphalopathies dégénératives et toxiques, notamment après emploi du méthotrexate ou du traitement par les radiations ionisantes.
· Allaitement (voir rubrique 4.6).
· Vaccins vivants atténués (contre fièvre jaune, varicelle - zona, rougeole, oreillons, rubéole, tuberculose, rotavirus, grippe) et ce pendant les 6 mois suivant l’arrêt de la chimiothérapie (voir rubrique 4.5) : risque de maladie vaccinale généralisée éventuellement mortelle.
4.4. Mises en garde spéciales et précautions d'emploi
La cytarabine est un puissant myélosuppresseur : elle peut entraîner une hypoplasie ou une aplasie médullaire dont la sévérité dépend de la dose administrée et du schéma thérapeutique utilisé.
Insuffisance médullaire préexistante : la cytarabine peut être administrée en cas de nécessité absolue.
Le traitement doit dans ce cas être initié avec prudence.
Les patients recevant ce traitement doivent être placés sous surveillance médicale stricte.
Pendant la phase d’induction une numération des globules blancs et des plaquettes doit être réalisée quotidiennement. Des examens médullaires doivent être réalisés fréquemment une fois que les cellules blastiques ont disparu du sang périphérique.
Il conviendra de considérer la possibilité de suspendre ou de modifier le traitement lorsque l’insuffisance médullaire médicamenteuse entraîne une réduction du nombre de plaquettes à moins de 50 000 ou de polynucléaires neutrophiles à moins de 1 000/mm3. Il se peut que le nombre d’éléments figurés continue à diminuer après l’arrêt du traitement pour atteindre les valeurs les plus basses après une période sans traitement de 12 à 24 jours. Si cela est indiqué, la reprise du traitement peut se faire lorsque des signes nets de réparation médullaire apparaissent.
Un équipement spécial doit être disponible afin de pouvoir gérer les complications, potentiellement fatales de l’insuffisance médullaire (infections résultant d’une granulopénie et autre diminution des défenses de l’organisme, hémorragies secondaires à la thrombopénie).
On surveillera les fonctions hépatiques et rénales. Les patients ayant une insuffisance hépatique ou rénale présentent un risque plus important de toxicité sur le système nerveux central après administration de fortes doses de cytarabine. Il faudra donc utiliser le produit avec précaution en réduisant les doses chez les patients atteints d’insuffisance hépatique et rénale.
Syndrome de lyse tumorale : comme toute chimiothérapie antileucémique, la cytarabine induit une hyperuricémie secondaire à la lyse cellulaire : on surveillera le taux d’acide urique pendant le traitement et on préviendra l’hyperuricémie.
Les patients recevant des doses élevées de cytarabine doivent être suivis afin de détecter des signes de neuropathie, car il peut être nécessaire de modifier le schéma d’administration et les doses pour éviter des troubles neurologiques irréversibles (voir rubrique 4.8).
La vaccination avec un vaccin vivant est contre-indiquée chez les patients recevant de la cytarabine (voir rubrique 4.5).
L’association de ce médicament est déconseillée avec la phénytoïne (et par extrapolation la fosphénytoïne) (voir rubrique 4.5).
Femmes en âge de procréer traitées (voir rubrique 4.6)
Les femmes en âge de procréer traitées par la cytarabine doivent utiliser un moyen de contraception efficace au cours du traitement et 6 mois après la fin du traitement.
Hommes traités (voir rubrique 4.6)
Il est souhaitable que les hommes traités par la cytarabine ou leur partenaire utilisent une méthode contraceptive de manière à éviter une conception pendant le traitement du patient et dans les 3 mois suivant la fin du traitement.
Les patients traités doivent être avertis de la nécessité de consulter en vue d’une conservation de sperme préalablement au traitement, en raison de la possibilité d’atteinte de la fertilité.
Excipient
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
En raison de l’augmentation du risque thrombotique lors des affections tumorales, le recours à un traitement anticoagulant est fréquent. La grande variabilité de la coagulabilité au cours de ces affections, à laquelle s’ajoute l’éventualité d’une interaction entre les anticoagulants oraux et la chimiothérapie anticancéreuse, impose, s’il est décidé de traiter le patient par anticoagulants oraux, d’augmenter la fréquence des contrôles de l’INR (acénocoumarol, fluindione, phénindione, tioclomarol, warfarine).
Interactions communes à tous les cytotoxiques
Association contre-indiquée (voir rubrique 4.3)
+ Vaccins vivants atténués (contre fièvre jaune, varicelle - zona, rougeole, oreillons, rubéole, tuberculose, rotavirus, grippe) et ce pendant les 6 mois suivant l’arrêt de la chimiothérapie :
Risque de maladie vaccinale généralisée éventuellement mortelle.
Associations déconseillées (voir rubrique 4.4)
+ Phénytoïne (et, par extrapolation, fosphénytoïne)
Risque de survenue de convulsions par diminution de l’absorption digestive de la seule phénytoïne par le cytotoxique ou bien risque de majoration de la toxicité ou de perte d’efficacité du cytotoxique par augmentation de son métabolisme hépatique par la phénytoïne ou la fosphénytoïne.
Associations faisant l'objet de précautions d’emploi
+ Antivitamines K
Augmentation du risque thrombotique et hémorragique au cours des affections tumorales. De surcroît, possible interaction entre les AVK et la chimiothérapie.
Contrôle plus fréquent de l'INR.
Association à prendre en compte
+ Immunosuppresseurs (ciclosporine, évérolimus, sirolimus, tacrolimus, temsirolimus)
Immunodépression excessive avec risque de syndrome lymphoprolifératif.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer/Contraception chez les hommes et les femmes
En raison du risque de génotoxicité, il faut conseiller aux femmes en âge de procréer d'utiliser une méthode de contraception très efficace pendant le traitement et pendant 6 mois après la dernière dose de cytarabine.
En raison du risque de génotoxicité, il faut conseiller aux patients de sexe masculin ayant des partenaires féminines en âge de procréer d’utiliser une contraception très efficace pendant le traitement et pendant 3 mois après la dernière dose de cytarabine.
Grossesse
Compte tenu des données disponibles, la cytarabine ne sera administrée pendant la grossesse que si la pathologie met en jeu le pronostic vital de la mère. En effet, les études sur les fonctions de reproduction réalisées chez différentes espèces animales ont montré que la cytarabine est embryotoxique et a des effets tératogènes principalement sur le cerveau et le squelette.
Quelques cas de malformations congénitales des membres et de l’oreille externe ont été rapportés lors de l’exposition au premier trimestre de grossesse. En cas d’exposition au premier trimestre, une surveillance échographique orientée est donc recommandée.
Des cas de prématurité ou de retard de croissance intra-utérin ont été signalés.
A la naissance, la survenue d’ictère, d’insuffisance médullaire et d’hyperéosinophilie transitoires a été rapportée. Une surveillance biologique est donc indiquée dans les premières semaines de vie.
Allaitement
L’excrétion de la cytarabine dans le lait maternel n’est pas connue. En raison des effets indésirables potentiellement graves pouvant être entraînés par la cytarabine chez les enfants allaités, la prise de cytarabine doit être contre-indiquée au cours de l’allaitement. L’allaitement doit être interrompu pendant le traitement par cytarabine et doit être évité pendant au moins deux jours après la dernière dose du traitement.
Fertilité
La cytarabine est mutagène et peut induire une atteinte chromosomique des spermatozoïdes.
Les patients traités doivent être avertis de la nécessité de consulter en vue d’une conservation de sperme préalablement au traitement, en raison de la possibilité d’atteinte de la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité d’emploi
Affections hématologiques et du système lymphatique
La cytarabine est un agent antinéoplasique qui entraîne une myélodépression. Son administration entraîne donc une aplasie ou une hypoplasie médullaire responsable d'anémie, granulopénie, thrombopénie, mégaloblastose et chute du taux de réticulocytes.
La sévérité de l'aplasie dépend de la dose administrée et du schéma thérapeutique utilisé. En relation avec l'aplasie, des complications hémorragiques ou infectieuses graves peuvent venir compliquer secondairement la cure de chimiothérapie.
Infections et infestations
Des infections virales, bactériennes, fongiques, parasitaires et saprophytiques peuvent être associées à l’utilisation de la cytarabine seule ou en association avec d’autres médicaments immunosuppresseurs affectant l’immunité cellulaire ou humorale. Ces infections peuvent être légères, mais elles peuvent aussi être graves et parfois fatales.
Affections respiratoires, thoraciques et médiastinales
De rares cas de pneumopathies interstitielles ont été rapportés chez des patients traités avec des doses intermédiaires de cytarabine associée ou non à d’autres agents de chimiothérapie, sans que cela ait pu être associé de façon claire à la cytarabine.
Affections gastro-intestinales
Les nausées et vomissements sont plus fréquents à la suite d’une perfusion rapide.
Des cas de pancréatite aiguë ont été rapportés chez des patients traités avec de la cytarabine en association avec d’autres médicaments.
Affections musculo-squelettiques et systémiques
Le syndrome cytarabine se caractérise par élévation thermique, myalgies, douleurs osseuses accompagnées dans certains cas par des douleurs thoraciques, rashs maculopapuleux, conjonctivite et sensation de malaise général. Ce syndrome survient 6 à 12 heures après l'administration du produit.
Son traitement et sa prévention répondent aux corticoïdes.
Investigations
Dans de rares cas, une hyperuricémie secondaire à la lyse blastique peut être induite par le traitement à la cytarabine ; il sera donc nécessaire de surveiller le taux d'acide urique dans le sang et les urines.
Les données de sécurité sont issues des bases de données internes de pharmacovigilance et des recherches bibliographiques.
Tableau récapitulatif des effets indésirables (doses conventionnelles et fortes doses)
Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Les fréquences des événements sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Infections et infestations |
|
|
Très fréquent |
Septicémie, pneumonie, infection a |
|
Fréquence indéterminée |
Cellulite au point d’injection |
|
Affections hématologiques et du système lymphatique |
|
|
Très fréquent |
Aplasie, insuffisance médullaire, thrombocytopénie, anémie, anémie mégaloblastique, leucopénie, neutropénie, diminution du taux de réticulocytes |
|
Affections du système immunitaire |
|
|
Fréquence indéterminée |
Réaction anaphylactique, œdème allergique |
|
Troubles du métabolisme et de la nutrition |
|
|
Fréquent |
Diminution de l’appétit |
|
Affections du système nerveux |
|
|
Fréquence indéterminée |
Neurotoxicité, névrite, étourdissements, maux de tête |
|
Affections oculaires |
|
|
Fréquence indéterminée |
Conjonctivite (voir syndrome cytarabine) b |
|
Affections cardiaques |
|
|
Fréquence indéterminée |
Péricardite, bradycardie sinusale |
|
Affections vasculaires |
|
|
Fréquence indéterminée |
Thrombophlébite |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Rare |
Pneumopathie interstitielle |
|
Fréquence indéterminée |
Dyspnée, douleur oropharyngée |
|
Affections gastro-intestinales |
|
|
Très fréquent |
Stomatite, ulcère buccal, ulcère anal, inflammation anale, diarrhée, vomissement, nausée, douleur abdominale, mucite |
|
Fréquence indéterminée |
Pancréatite, ulcère de l’œsophage, œsophagite |
|
Affections hépatobiliaires |
|
|
Très fréquent |
Fonction hépatique anormale |
|
Fréquence indéterminée |
Ictère |
|
Affections de la peau et du tissu sous-cutané |
|
|
Très fréquent |
Alopécie, rash |
|
Fréquent |
Ulcère cutané |
|
Fréquence indéterminée |
Syndrome d’érythrodysesthésie palmo-plantaire, urticaire, prurit, éphélides, dermite exfoliative, hidradénite neutrophilique eccrine, érythème auriculaire (érythème de l’oreille lié à la cytarabine) |
|
Affections musculo-squelettiques et systémiques |
|
|
Très fréquent |
Syndrome cytarabine |
|
Affections du rein et des voies urinaires |
|
|
Fréquence indéterminée |
Insuffisance rénale, rétention urinaire |
|
Affections des organes de reproduction et du sein |
|
|
Fréquence indéterminée |
Aménorrhée, azoospermie |
|
Troubles généraux et anomalies au site d'administration |
|
|
Très fréquent |
Pyrexie |
|
Fréquence indéterminée |
Réaction au site d’injection c |
|
Investigations |
|
|
Très fréquent |
Biopsie de la moelle osseuse anormale, frottis sanguin anormal |
|
Rare |
Hyperuricémie |
|
a Infections virales, bactériennes, fongiques et parasitaires, parfois mortelles. b Peut apparaître avec une éruption et peut être hémorragique en cas de thérapie à forte dose. c Douleur et inflammation au site d'injection sous-cutanée. |
|
Description d’effets indésirables particuliers
Effets indésirables associés à la voie intrathécale
Les effets les plus fréquemment rapportés après administration par voie intrathécale sont des nausées, des vomissements et de la fièvre. Ces réactions sont légères.
Des accidents de neurotoxicité graves dont des paraplégies ont été rapportés lors d’administrations intrathécales combinées avec du méthotrexate et des corticostéroïdes et lors d’association d’injection intrathécale avec une administration systémique de fortes doses de méthotrexate et de cytarabine.
Des cas de leucoencéphalites nécrosantes avec ou sans convulsion ont été rapportés. Certains de ces patients ont aussi été traités par méthotrexate et/ou hydrocortisone par voie intrathécale et par irradiation encéphalique.
Deux cas de cécité ont été décrits chez des sujets mis en rémission après polychimiothérapie intraveineuse et traitement préventif des greffes méningées avec cytarabine intrathécale et radiothérapie de l'encéphale.
Effets indésirables associés aux fortes doses
Affections du système nerveux
Toxicité neurologique à forte dose.
Atteintes cérébelleuses de formes légère (dysarthrie et nystagmus) à grave (ataxie sévère pouvant être d’apparition retardée et dans certains cas définitive). Des épisodes de comas et des neuropathies périphériques sensitives et motrices ont aussi été rapportés. Des effets graves à mortels ont été observés chez des malades ayant antérieurement reçu d’autres traitements sur le système nerveux central (irradiation encéphalique) : il est recommandé de ne pas dépasser la dose individuelle recommandée et d’être très prudent chez les patients ayant déjà été traités par radiothérapie ou par voie intrathécale.
La toxicité neurologique semble associée au débit rapide d’administration.
Affections oculaires
Des atteintes réversibles de la cornée et des conjonctivites hémorragiques ont été décrites après utilisation de fortes doses de cytarabine. Ces effets peuvent être prévenus ou diminués par l'instillation d'un collyre contenant des corticoïdes.
Affections cardiaques
Des cas de cardiomyopathie pouvant être fatals ont été rapportés suite à l’utilisation expérimentale d’un traitement utilisé dans le cadre de transplantation médullaire, associant de fortes doses de cytarabine à du cyclophosphamide.
Affections respiratoires, thoraciques et médiastinales
Une toxicité pulmonaire sévère, parfois fatale, un syndrome de détresse respiratoire et un œdème pulmonaire ont été rapportés après utilisation de fortes doses de cytarabine.
Tableau récapitulatif des effets indésirables (fortes doses seulement)
Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Les fréquences des événements sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Infections et Infestations |
|
|
Fréquence indéterminée |
Abcès hépatique |
|
Affections du système nerveux |
|
|
Très fréquent |
Trouble cérébral, trouble cérébelleux, somnolence |
|
Fréquence indéterminée |
Coma, convulsion, neuropathie motrice périphérique, neuropathie sensitive périphérique |
|
Affections oculaires |
|
|
Très fréquent |
Affection de la cornée |
|
Affections cardiaques |
|
|
Fréquence indéterminée |
Cardiomyopathie a |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Très fréquent |
Syndrome de détresse respiratoire aiguë, œdème pulmonaire, toxicité pulmonaire |
|
Affections gastro-intestinales |
|
|
Fréquent |
Colite nécrosante |
|
Fréquence indéterminée |
Nécrose gastro-intestinale, pneumatose de l’intestin, péritonite |
|
Affections hépatobiliaires |
|
|
Fréquence indéterminée |
Atteinte hépatique, hyperbilirubinémie |
|
Affections de la peau et du tissu sous-cutané |
|
|
Fréquent |
Desquamation cutanée |
|
Fréquence indéterminée |
Hidradénite neutrophilique eccrine, érythème auriculaire (érythème de l’oreille lié à la cytarabine) |
|
a Pouvant entraîner le décès. |
|
Population pédiatrique
Le profil des effets indésirables de la cytarabine a été similaire dans la population pédiatrique par rapport aux adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Antimétabolite spécifique de la phase S du cycle cellulaire (phase de division cellulaire).
La cytotoxicité de la cytarabine dépend de son métabolite actif l'ARA-CTP qui incorporé à l'ADN en bloque la synthèse. La molécule d’ADN comprenant de l'ARA-CTP présente des anomalies structurales aboutissant à des perturbations du métabolisme cellulaire et altérant sa reproduction. La cytotoxicité passerait aussi par une inhibition de l'ADN polymérase et par une action sur le système des kinases.
L'utilisation de hautes doses de cytarabine a montré qu'elles permettent de vaincre la résistance des cellules leucémiques ne répondant plus aux doses conventionnelles du produit.
Plusieurs mécanismes semblent intervenir pour vaincre cette résistance :
· augmentation de la quantité de substrat,
· augmentation du pool intracellulaire d'ARA-CTP : il existe une corrélation positive entre la rétention intracellulaire d'ARA-CTP et le pourcentage de cellules en phase S.
5.2. Propriétés pharmacocinétiques
Après administration intraveineuse d'une dose de 2 à 3 g/m² toutes les 12 heures en perfusion d'une heure sur 5 à 6 jours (10 à 12 doses), les concentrations plasmatiques en fin de perfusion sont de l'ordre de : 19,96 + 8,02 µg/ml et 35 + 2,8 µg/ml. Les concentrations plasmatiques décroissent à l'arrêt de la perfusion, selon une courbe biexponentielle. Six heures après la fin de la perfusion, les concentrations obtenues correspondent à celles mesurées au « steady-state » après une perfusion continue de 24 heures de 100 mg/m² de cytarabine.
Par comparaison avec la cinétique de la cytarabine à dose conventionnelle, les hautes doses produisent un pic 200 fois supérieur.
De même le pic d'apparition d'ARA-U métabolite inactif est retardé avec les hautes doses puisqu'il n'apparaît qu'au bout de 15 minutes.
Aux doses conventionnelles :
· la T½ A est de l'ordre de quelques minutes (10 en moyenne),
· la T½ B est de l'ordre de quelques heures (1 à 3).
Liaison aux protéines : 14 % de la cytarabine environ est liée aux protéines plasmatiques.
Clairance rénale plus lente avec les hautes doses, de l'ordre de 232 + 33,4 ml/min/m².
La cytarabine administrée par voie générale (I.V.) passe la barrière hémato-encéphalique : après une dose de 1 à 3 g/m² en perfusion de 1 à 3 heures, les concentrations dans le liquide céphalo-rachidien sont de l'ordre de 100 à 300 ng/ml.
Le produit diffuse aussi dans la salive, la rate, les reins, le tube digestif, le thymus, la moelle osseuse et les larmes. On ne sait pas si la cytarabine passe dans le lait maternel.
Activation de la cytarabine en ARA-CTP métabolite actif
Passage de la membrane cellulaire par une diffusion facilitée selon le gradient de concentration à haute concentration, par un mécanisme utilisant un transporteur à faible concentration.
Activation enzymatique par phosphorylations successives : les enzymes qui activent l'ARA-C sont celles qui assurent l'activation du ribonucléoside naturel, la déoxycytidine.
Deux enzymes jouent un rôle important : déoxycytidine kinase (ARA-C ® ARA-CMP) et déoxycytidilate kinase (ARA-CMP ® ARA-CDP).
Le métabolite actif formé est l'ARA-CTP (arabinofuranosylcytosine triphosphate). La formation de l'ARA-CTP est une condition nécessaire à la cytotoxicité du produit mais n'est semble-t-il pas la seule : d'autres mécanismes interviennent.
Catabolisme
La cytarabine est dégradée en ARA-U (arabinofuranosyl uracile), métabolite inactif, par la cytidine déaminase, enzyme présente dans de nombreux tissus mais principalement dans le foie et aussi dans les cellules leucémiques et la moelle. Cette enzyme est la cible de nombreux phénomènes d'activation ou d'inhibition.
5.3. Données de sécurité préclinique
Aucune étude de fertilité n'a été réalisée, mais des effets sur la fertilité mâle ont été rapportés chez la souris. La cytarabine est embryotoxique et tératogène (cerveau et squelette) et est responsable d’une toxicité péri- et post-natale chez de nombreuses espèces. Administrée à des rats nouveau-nés à la dose de 4mg/kg/j, la cytarabine a provoqué des retards de développement.
La cytarabine est mutagène et clastogène.
Aucune étude de cancérogenèse n'a été réalisée.
Ce médicament ne doit pas être mélangé avec d’autres médicaments à l’exception de ceux mentionnés dans la rubrique 4.2. S’assurer de la compatibilité avant de le mélanger ou de l’associer à toute autre substance.
Après ouverture/reconstitution/dilution : le produit doit être utilisé immédiatement.
6.4. Précautions particulières de conservation
Pas de précaution particulière de conservation.
6.5. Nature et contenu de l'emballage extérieur
1 g en flacon (verre) ; boîte de 1 ou 5 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
La manipulation de ce cytotoxique par le personnel infirmier ou médical nécessite un ensemble de précautions permettant d’assurer la protection du manipulateur et de son environnement (voir rubrique 4.2).
Ne pas utiliser une solution dans laquelle un léger trouble serait apparu.
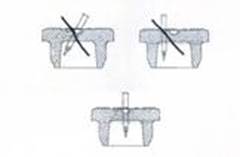
La reconstitution doit s'effectuer à l'aide d'une seringue munie d'une aiguille d'un diamètre extérieur de 0,8 mm (équivalent à 21 Gauges). L'utilisation d'une aiguille de diamètre supérieur risquerait d'entraîner la chute du bouchon ou de fragments de bouchon dans le flacon.
A l’aide de l’aiguille, percez au centre de l’opercule de façon perpendiculaire au bouchon selon le schéma donné ci-dessous :
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
23-25 AVENUE DU DOCTEUR LANNELONGUE
75014 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 562 016 7 9 : 1 g en flacon (verre) ; boîte de 1.
· 34009 555 784 2 0 : 1 g en flacon (verre) ; boîte de 5.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Réservé à l’usage hospitalier.
Réservé aux spécialistes en oncologie, en hématologie et en médecine interne.
ANSM - Mis à jour le : 14/04/2026
ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.)
Cytarabine
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) ?
3. Comment utiliser ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) ET DANS QUELS CAS EST-IL UTILISE ?
Ce médicament empêche la croissance de certaines cellules. Il est utilisé dans le traitement de certaines maladies du sang.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) ?
N’utilisez jamais ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) :
· si vous êtes allergique à la cytarabine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6,
· si vous allaitez,
· si vous êtes atteint d’encéphalopathies dégénératives et toxiques (affections neurologiques), notamment après l’emploi du méthotrexate ou de traitement par les radiations ionisantes,
· en cas d’aplasie médullaire préexistante (appauvrissement de la moelle osseuse en cellules sanguines),
· en association avec les vaccins vivants atténués (vaccins contre la fièvre jaune, la varicelle, le zona, la rougeole, les oreillons, la rubéole, la tuberculose, le rotavirus, la grippe) et ce pendant les 6 mois suivant l’arrêt de la chimiothérapie (voir « Autres médicaments et ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) »).
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) :
· le traitement ne peut être administré que sous SURVEILLANCE MEDICALE RIGOUREUSE,
· celle-ci comporte habituellement un examen clinique et des examens biologiques notamment numération formule sanguine, myélogramme (examen de la moelle osseuse), taux d’acide urique,
· les fonctions hépatique et rénale seront aussi surveillées,
· si vous avez reçu un traitement radiothérapique, n’omettez pas de le signaler à votre médecin,
· si vous présentez une insuffisance médullaire, ce médicament ne vous sera administré qu’en cas de nécessité absolue et des examens supplémentaires seront réalisés en particulier au début du traitement,
· si vous devez vous faire vacciner, vous devez impérativement prévenir votre médecin traitant que vous suivez un traitement avec ARACYTINE,
· ce médicament doit être évité en association avec la phénytoïne (et, par extrapolation, la fosphénytoïne (utilisés dans le traitement de l’épilepsie).
Si vous désirez avoir un enfant
ARACYTINE peut altérer la fertilité masculine. Parlez à votre médecin de la préservation de la fertilité avant de commencer le traitement. Les hommes et les femmes doivent utiliser des mesures contraceptives efficaces (voir rubrique « Grossesse, allaitement et fertilité »).
Enfants
Sans objet.
Autres médicaments et ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.)
Ce médicament NE DOIT PAS ETRE UTILISE en association avec :
· les vaccins vivants atténués (vaccins contre la fièvre jaune, la varicelle, le zona, la rougeole, les oreillons, la rubéole, la tuberculose, le rotavirus, la grippe) et ce pendant les 6 mois suivant l’arrêt de la chimiothérapie.
Ce médicament DOIT ETRE EVITE en association avec :
· la phénytoïne (et, par extrapolation, la fosphénytoïne) (médicaments utilisés dans le traitement de l’épilepsie).
Prévenez votre médecin si vous prenez des médicaments de la classe des antivitamines K ou des immunosuppresseurs.
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Grossesse
Evitez de débuter une grossesse pendant votre traitement par ARACYTINE car il existe un risque de malformations congénitales. Si vous débutez une grossesse, vous devez en informer immédiatement votre médecin.
Contraception chez les femmes en âge de procréer
Les femmes en âge d’avoir des enfants doivent réaliser un test de grossesse avant de commencer le traitement par ARACYTINE et toujours utiliser une contraception efficace qui leur sera prescrite par leur médecin afin d’éviter une grossesse au cours du traitement et 6 mois après la fin du traitement. Discutez avec votre médecin des méthodes de contraception efficaces pour vous et votre partenaire.
Les hommes traités par ARACYTINE ayant des partenaires en âge d’avoir des enfants doivent toujours utiliser une contraception efficace afin d’éviter une grossesse pendant le traitement et dans les 3 mois suivant la fin du traitement.
Allaitement
Ce médicament est contre-indiqué pendant l’allaitement. L’allaitement doit être interrompu pendant le traitement par ARACYTINE et doit être évité pendant au moins deux jours après la dernière dose du traitement.
Fertilité
Conduite de véhicules et utilisation de machines
Soyez très prudent : Ne pas conduire sans l’avis d’un professionnel de santé.
ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) ?
La dose administrée doit être évaluée en tenant compte de l’état clinique du patient (hématologique, extra-hématologique, hépatique et rénal).
Le traitement doit être adapté à chaque cas.
Mode et voie d’administration
Elle est administrée en perfusion intraveineuse dans 250 ml de solution isotonique de glucose ou de solution isotonique de chlorure de sodium d’une durée de 1 à 3 heures.
Ne pas utiliser de solvant contenant de l’alcool benzylique.
A l'attention du personnel soignant
Comme pour tout cytotoxique, la préparation et la manipulation de ce produit nécessitent un ensemble de précautions permettant d'assurer la protection du manipulateur et de son environnement, dans les conditions de sécurité requises pour le patient.
En plus des précautions usuelles pour préserver la stérilité des préparations injectables, il faut :
· mettre une blouse à manches longues et poignets serrés, afin d'éviter toute projection de solution sur la peau,
· porter également un masque chirurgical à usage unique et des lunettes enveloppantes,
· mettre des gants à usage unique en PVC, et non en latex, après lavage aseptique des mains,
· préparer la solution sur un champ de travail,
· arrêter la perfusion, en cas d'injection hors de la veine,
· éliminer tout matériel ayant servi à la préparation de la solution (seringues, compresses, champs, flacon) dans un conteneur réservé à cet effet,
· détruire les déchets toxiques,
· manipuler les excréta et vomissures avec précaution.
Les femmes enceintes doivent éviter la manipulation des cytotoxiques.
Attention
Il est extrêmement important de s’assurer que l’administration est intraveineuse. Toute extravasation risquerait de produire une nécrose des tissus environnants. En cas d’extravasation, l’administration sera interrompue immédiatement.
Incompatibilités
Il existe une incompatibilité physico-chimique de la cytarabine avec l'héparine, l'insuline, le 5 fluoro-uracile, la nafcilline, l'oxacilline, la pénicilline G, le solu-B (solution injectable de vitamines du groupe B, de vitamines C et PP) et l’hémisuccinate de méthylprednisolone.
Si vous avez utilisé plus de ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) que vous n’auriez dû
Il n'existe pas d'antidote spécifique. La dose de 4,5 g/m² en perfusion IV d'une heure toutes les 12 heures en 12 doses, provoque une toxicité du système nerveux central irréversible et létale.
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez d’utiliser ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.)
Sans objet.
Si vous arrêtez d’utiliser ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.)
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Au cours du traitement, il est possible que surviennent :
Des effets indésirables très fréquents (pouvant apparaître chez plus de 1 patient sur 10)
Des infections :
· infection généralisée (septicémie),
· infection des poumons (pneumonie),
· autres infections (pouvant être virales, bactériennes, fongiques, parasitaires et dans certains cas mortelles).
Des modifications du bilan sanguin :
· diminution des globules blancs dans le sang (leucopénie) ou diminution de certains globules blancs dans le sang (neutropénie), pouvant s’accompagner de frissons et de fièvre, qui nécessitent immédiatement un avis médical. Elle peut être responsable d’infection,
· diminution des plaquettes sanguines (thrombocytopénie) pouvant s’accompagner de saignements qui nécessitent immédiatement un avis médical,
· diminution des globules rouges (anémie, anémie mégaloblastique, diminution du taux de réticulocytes),
· appauvrissement de la moelle osseuse en cellules sanguines (aplasie) ou défaillance de la production de ces cellules par la moelle osseuse (insuffisance médullaire).
Des effets digestifs :
· nausées, vomissements, diarrhée,
· ulcération ou inflammation (stomatite) au niveau de la bouche,
· ulcération ou inflammation au niveau anal,
· inflammation des muqueuses (mucite),
· douleurs abdominales.
Des effets au niveau du foie :
· fonctionnement du foie anormal.
Des effets au niveau de la peau :
· rash, perte des cheveux.
Des effets généraux :
· le syndrome cytarabine peut apparaître 6 à 12 heures après l'administration du produit, se caractérise par un ensemble de troubles associant température, douleurs musculaires et osseuses, parfois douleurs de la poitrine, inflammation des conjonctives, éruption cutanée et sensation de malaise général. Ces troubles peuvent dans certains cas être atténués et même prévenus par l'administration de médicaments contenant des corticoïdes ;
· fièvre (pyrexie).
Des effets au niveau des analyses biologiques :
· biopsie de la moelle osseuse anormale, frottis sanguin anormal.
Des effets indésirables fréquents (pouvant apparaître chez 1 à 10 patients sur 100)
Des effets sur le métabolisme et la nutrition :
· diminution de l’appétit.
Des effets au niveau de la peau :
· ulcère de la peau.
Des effets indésirables rares (pouvant apparaître chez 1 à 10 patients sur 10 000)
Des effets pulmonaires :
· infection pulmonaire (pneumopathie interstitielle).
Des effets au niveau des analyses biologiques :
· augmentation de l’acide urique dans le sang (hyperuricémie).
Des effets indésirables de fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles)
Des infections :
· inflammation d’une couche profonde de la peau (cellulite) au point d’injection.
Des effets sur le système immunitaire :
· réactions allergiques graves (réactions anaphylactiques),
· œdème allergique.
Des effets sur le système nerveux :
· neurotoxicité,
· inflammation d’un nerf (névrite),
· étourdissements,
· maux de tête.
Des effets au niveau des yeux :
· inflammation de la conjonctive (conjonctivite – se référer ci-dessus au syndrome cytarabine).
Des effets cardiaques :
· inflammation de l’enveloppe du cœur (péricardite), ralentissement de la fréquence cardiaque (bradycardie sinusale).
Des effets vasculaires :
· inflammation des veines dans lesquelles se forme un caillot (thrombophlébite).
Des effets pulmonaires :
· difficulté à respirer (dyspnée), douleur oropharyngée.
Des effets digestifs :
· ulcération de l’œsophage,
· inflammation de l’œsophage (œsophagite),
· inflammation du pancréas (pancréatite).
Des effets au niveau du foie :
· jaunisse (ictère).
Des effets au niveau de la peau :
· éruption cutanée des paumes des mains et plantes des pieds (érythrodysesthésie palmo-plantaire),
· urticaire,
· démangeaison (prurit),
· taches brunes (éphélides),
· inflammation de la peau accompagnée de desquamation (dermite exfoliative),
· rougeur, douleur ou gonflement des oreilles pouvant survenir pendant ou peu de temps après le traitement par cytarabine (érythème auriculaire ou érythème de l’oreille lié à la cytarabine),
· inflammation des glandes sudoripares, causant parfois des tâches rouges sensibles sur la peau (hidradénite neutrophilique eccrine).
Des effets sur le rein :
· défaillance des fonctions rénales (insuffisance rénale),
· difficulté à évacuer l’urine (rétention urinaire).
Des effets sur la reproduction :
· absence de règles (aménorrhée),
· absence de spermatozoïdes dans le sperme (azoospermie).
Des effets généraux et au site d’injection :
· réaction au site d’injection (douleur et inflammation).
Au cours du traitement avec de fortes doses seulement, il est possible que surviennent :
Des effets indésirables très fréquents (pouvant apparaître chez plus de 1 patient sur 10)
Des effets sur le système nerveux :
· troubles au niveau de cerveau,
· troubles au niveau du cervelet pouvant être d’intensité légère, se manifestant par des troubles de la prononciation (dysarthrie) ou des mouvements involontaires des yeux (nystagmus), à grave, se manifestant par une difficulté à coordonner les mouvements (ataxie sévère parfois d’apparition retardée et dans certains cas définitive),
· somnolence.
Des effets au niveau des yeux :
· affection de la cornée.
Des effets pulmonaires pouvant dans certains cas être graves, parfois fatals :
· syndrome de détresse respiratoire aiguë,
· œdème pulmonaire,
· toxicité pulmonaire.
Des effets indésirables fréquents (pouvant apparaître chez 1 à 10 patients sur 100)
Des effets digestifs :
· inflammation du côlon (colite nécrosante).
Des effets au niveau de la peau :
· desquamation cutanée.
Des effets indésirables de fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles)
Au niveau du foie :
· abcès du foie.
Au niveau du système nerveux :
· coma,
· convulsions,
· sensation de faiblesse au niveau des membres (neuropathie motrice périphérique),
· perte de la sensibilité au toucher ou troubles de la sensibilité (fourmillements, picotements, sensations douloureuses, sensation de chaud ou de froid) au niveau des membres (neuropathie sensitive périphérique).
Au niveau cardiaque :
· maladie du muscle cardiaque (cardiomyopathie) pouvant entraîner la mort.
Au niveau digestif :
· nécrose gastro-intestinale,
· groupe de kystes gazeux dans l’épaisseur de la paroi de l’intestin (pneumatose de l’intestin),
· inflammation de l’abdomen (péritonite).
Au niveau hépatobiliaire :
· atteinte du foie,
· augmentation du taux sanguin de bilirubine.
Des effets au niveau de la peau :
· rougeur, douleur ou gonflement des oreilles pouvant survenir pendant ou peu de temps après le traitement par cytarabine (érythème auriculaire ou érythème de l’oreille lié à la cytarabine),
· inflammation des glandes sudoripares, causant parfois des tâches rouges sensibles sur la peau (hidradénite neutrophilique eccrine).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance – Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.) ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le conditionnement extérieur. La date de péremption fait référence au dernier jour de ce mois.
Ne pas utiliser une solution dans laquelle un léger trouble serait apparu.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ARACYTINE 1 g, poudre pour solution pour perfusion (I.V.)
· La substance active est :
cytarabine............................................................................................................................ 1 g
Pour un flacon de poudre.
Ce médicament se présente sous la forme d’une poudre pour solution pour perfusion (I.V.). Boîte de 1 ou 5 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
PFIZER HOLDING FRANCE
23-25 AVENUE DU DOCTEUR LANNELONGUE
75014 PARIS
Exploitant de l’autorisation de mise sur le marché
PFIZER
23-25 AVENUE DU DOCTEUR LANNELONGUE
75014 PARIS
LATINA PHARMA S.p.A.
VIA MURILLO, 7
04013 SERMONETA (LT)
ITALIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
A compléter ultérieurement par le titulaire
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Avant utilisation, la cytarabine peut être reconstituée avec le solvant ci-dessous :
· eau pour préparation injectable.
Les volumes de solvant à utiliser pour la reconstitution sont comme suit :
· cytarabine 500 mg est reconstituée avec 10 ml de solvant,
· cytarabine 1 g est reconstituée avec 10 ml de solvant,
· cytarabine 2 g est reconstituée avec 20 ml de solvant.
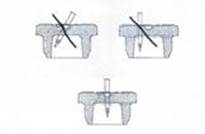
La reconstitution doit s'effectuer à l'aide d'une seringue munie d'une aiguille d'un diamètre extérieur de 0,8 mm (équivalent à 21 Gauges). L'utilisation d'une aiguille de diamètre supérieur risquerait d'entraîner la chute du bouchon ou de fragments de bouchon dans le flacon.
A l’aide de l’aiguille, percez au centre de l’opercule de façon perpendiculaire au bouchon selon le schéma donné ci-dessous :