Dernière mise à jour le 03/08/2026
ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé
Indications thérapeutiques
Classe pharmacothérapeutique : agent antinéoplasique inhibiteur de protéine kinase - code ATC: L01EB02.
ERLOTINIB BIOGARAN contient une substance active appelée erlotinib. ERLOTINIB BIOGARAN est un médicament destiné à traiter le cancer en bloquant l’activité d’une protéine appelée récepteur du facteur de croissance épidermique (EGFR). On sait que cette protéine intervient dans la croissance et la dissémination des cellules cancéreuses.
ERLOTINIB BIOGARAN est indiqué chez l’adulte. Ce médicament peut vous être prescrit si vous souffrez d’un cancer du poumon non à petites cellules à un stade avancé. Il peut vous être prescrit en traitement initial ou en traitement si votre maladie reste inchangée après une chimiothérapie initiale sous réserve que les cellules de votre cancer aient des mutations de l’EGFR spécifiques. Il peut également vous être prescrit si la progression n’a pas été arrêtée par une chimiothérapie précédente. Ce médicament peut vous être également prescrit en association à un autre médicament appelé gemcitabine si vous souffrez d’un cancer du pancréas à un stade métastatique.
Présentations
> plaquette(s) aluminium OPA : polyamide orienté PVC de 30 comprimé(s)
Code CIP : 34009 301 115 3 6
Déclaration de commercialisation : 30/03/2020
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 850,02 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 851,04 €
- Taux de remboursement :100 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : BIOGARAN
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription hospitalière
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux spécialistes et services HEMATOLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 014 365 4
ANSM - Mis à jour le : 16/08/2023
ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
sous forme de chlorhydrate d'erlotinib
Pour un comprimé pelliculé.
Excipient(s) à effet notoire : chaque comprimé pelliculé contient 143,90 mg de lactose sous forme monohydratée.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé blanc à jaunâtre, rond, biconvexe, pelliculé, avec gravure « 150 » sur une face, de diamètre 10,5 mm ± 5 %.
4.1. Indications thérapeutiques
Cancer Bronchique Non à Petites Cellules (CBNPC) :
ERLOTINIB BIOGARAN, est indiqué en première ligne de traitement des formes localement avancées ou métastatiques du cancer bronchique non à petites cellules (CBNPC) chez les patients présentant des mutations activatrices de l’EGFR.
ERLOTINIB BIOGARAN est également indiqué dans le traitement de switch maintenance des formes localement avancées ou métastatiques du CBNPC chez les patients avec mutations activatrices de l’EGFR et présentant une maladie stable après une première ligne de chimiothérapie.
ERLOTINIB BIOGARAN, est également indiqué dans le traitement des formes localement avancées ou métastatiques du CBNPC après échec d'au moins une ligne de chimiothérapie. Chez les patients avec des tumeurs sans mutations activatrices de l’EGFR, ERLOTINIB BIOGARAN est indiqué lorsque les autres options de traitement ne sont pas considérées appropriées.
Lors de la prescription de ERLOTINIB BIOGARAN, les facteurs associés à une survie prolongée doivent être pris en considération.
Aucun bénéfice en survie ou autres effets cliniquement significatifs du traitement n’ont été démontrés chez les patients dont l’expression du récepteur au facteur de croissance épidermique (EGFR) de la tumeur (déterminée par IHC) était négative (voir rubrique 5.1).
Cancer du pancréas :
ERLOTINIB BIOGARAN, en association à la gemcitabine, est indiqué dans le traitement du cancer du pancréas métastatique. Lors de la prescription de ERLOTINIB BIOGARAN, les facteurs associés à une survie prolongée doivent être pris en considération (voir rubriques 4.2 et 5.1). Aucun avantage en survie n’a été montré chez les patients ayant une maladie localement avancée.
4.2. Posologie et mode d'administration
Le traitement par ERLOTINIB BIOGARAN, doit être supervisé par un médecin expérimenté dans l'utilisation des traitements anticancéreux.
Patients atteints d’un Cancer Bronchique Non à Petites Cellules :
La recherche de mutation de l’EGFR doit être effectuée selon les indications approuvées (voir rubrique 4.1).
La posologie quotidienne recommandée de ERLOTINIB BIOGARAN est de 150 mg à prendre au moins une heure avant ou deux heures après la prise de nourriture.
Patients atteints d’un cancer du pancréas :
La posologie quotidienne recommandée de ERLOTINIB BIOGARAN est de 100 mg à prendre au moins une heure avant ou deux heures après la prise de nourriture, en association à la gemcitabine (voir le résumé des caractéristiques du produit de la gemcitabine dans l’indication cancer du pancréas). Chez les patients qui ne développent pas d’éruptions cutanées dans les 4 à 8 premières semaines de traitement, la poursuite du traitement par ERLOTINIB BIOGARAN doit être réévaluée (voir rubrique 5.1).
Quand une adaptation de la posologie est nécessaire, la dose doit être réduite par paliers de 50 mg (voir rubrique 4.4).
ERLOTINIB BIOGARAN est disponible en dosages de 25 mg, 100 mg et 150 mg.
L’administration concomitante de substrats et de modulateurs du CYP3A4 peut nécessiter une adaptation de la dose (voir rubrique 4.5).
Insuffisance hépatique :
L’erlotinib est éliminé par métabolisme hépatique et excrétion biliaire. Bien que l’exposition à l’erlotinib était similaire chez les patients ayant une insuffisance hépatique modérée (score de Child-Pugh 7-9) par rapport aux patients ayant une fonction hépatique adéquate, ERLOTINIB BIOGARAN, devra être utilisé avec précautions chez les patients présentant une insuffisance hépatique. Une réduction de la posologie ou une interruption de ERLOTINIB BIOGARAN, devrait être envisagée en cas de survenue d’effets indésirables graves. La tolérance et l’efficacité de l’erlotinib n’ont pas été étudiées chez les patients présentant un trouble hépatique sévère (ASAT/SGOT et ALAT/SGPT > 5 fois la limite supérieure de la normale). L’utilisation de ERLOTINIB BIOGARAN, chez les patients ayant un trouble hépatique sévère n’est pas recommandée (voir rubrique 5.2).
Insuffisance rénale :
La tolérance et l’efficacité de l’erlotinib n’ont pas été étudiées chez les patients insuffisants rénaux (créatinémie >1,5 fois la limite supérieure de la normale). Sur la base des données de pharmacocinétique, aucune adaptation de la posologie ne semble nécessaire chez les patients ayant une insuffisance rénale légère ou modérée (voir rubrique 5.2). L’utilisation de ERLOTINIB BIOGARAN, n’est pas recommandée chez les patients ayant une insuffisance rénale sévère.
Population pédiatrique :
La tolérance et l’efficacité de l’erlotinib dans les indications approuvées n’ont pas été établies chez des patients âgés de moins de 18 ans. L’utilisation de ERLOTINIB BIOGARAN, en pédiatrie n’est pas recommandée.
Fumeurs :
Il a été montré que le tabagisme réduit l’exposition à l’erlotinib de 50-60 %. La dose maximale tolérée de ERLOTINIB BIOGARAN chez les patients ayant un CBNPC et fumant des cigarettes est de 300 mg. La posologie de 300 mg n’a pas montré d’amélioration de l’efficacité en deuxième ligne de traitement après échec d’une chimiothérapie comparé à la posologie recommandée de 150 mg chez les patients qui continuent à fumer des cigarettes. Les données de tolérance étaient comparables entre la posologie de 300 mg et de 150 mg. Cependant, il y a eu une augmentation de l’incidence des éruptions cutanées, des affections pulmonaires interstitielles et des diarrhées, chez les patients recevant la posologie la plus élevée d’erlotinib. Les fumeurs devront être encouragés à arrêter de fumer (voir les rubriques 4.4, 4.5, 5.1 et 5.2).
Hypersensibilité à l’erlotinib ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Evaluation du statut de la mutation de l’EGFR
Lorsque l’utilisation de ERLOTINIB BIOGARAN en première ligne de traitement ou en traitement de maintenance des formes localement avancées ou métastatiques du cancer bronchique non à petites cellules (CBNPC) est envisagée, il est important que le statut de la mutation de l’EGFR d’un patient soit déterminé.
Un test validé, robuste, fiable et sensible, avec un seuil de positivité prédéfini et avec une utilité démontrée pour la détermination du statut de la mutation de l’EGFR, utilisant soit l’ADN tumoral provenant d’un échantillon de tissu ou l’ADN tumoral circulant (ADNtc) obtenu à partir d’un échantillon de sang (plasma), doit être réalisé selon les pratiques médicales locales.
Si un test d’ADNtc plasmatique est utilisé et que le résultat est négatif pour les mutations activatrices, un test tissulaire doit être réalisé chaque fois que possible en raison du risque de faux négatifs associé au test plasmatique.
Fumeurs
Les fumeurs devront être encouragés à arrêter de fumer, compte tenu de la réduction des concentrations plasmatiques d’erlotinib chez les fumeurs par rapport aux non-fumeurs. Le degré de réduction est probablement cliniquement significatif (voir les rubriques 4.2, 4.5, 5.1 et 5.2).
Affections pulmonaires interstitielles
Peu fréquemment, des évènements à type d’affections pulmonaires interstitielles (API), dont certains fatals, ont été décrits chez des patients traités par de l’erlotinib pour un cancer bronchique non à petites cellules (CBNPC), un cancer du pancréas ou d’autres tumeurs solides à un stade avancé. Au cours de l’étude pivot BR.21 dans le CBNPC, l’incidence des cas d’API (0,8 %) a été identique dans les groupes erlotinib et placebo. Dans une méta-analyse d’essais cliniques contrôlés randomisés dans le CBNPC (excluant les études de phase I et de phase II monobras en raison de l’absence de groupes contrôles), l’incidence des évènements à type d’API était de 0,9 % sous erlotinib comparée à 0,4 % chez les patients dans les bras contrôles. Lors de l’étude menée dans le cancer du pancréas en association à la gemcitabine, l’incidence des événements à type d’API était de 2,5 % dans le groupe erlotinib plus gemcitabine contre 0,4 % dans le groupe gemcitabine plus placebo. Chez les patients avec suspicion d’événements à type d’API, les diagnostics reportés incluaient notamment : pneumopathie inflammatoire, pneumopathie radique, pneumopathie d’hypersensibilité, pneumonie interstitielle, affection pulmonaire interstitielle, bronchiolite obstructive, fibrose pulmonaire, Syndrome de Détresse Respiratoire Aiguë (SDRA), alvéolite inflammatoire et infiltration pulmonaire. Les symptômes sont survenus quelques jours voire plusieurs mois après l’instauration du traitement par l’erlotinib. La plupart des cas ont été fréquemment associés à des facteurs confondants ou favorisants tels qu'une chimiothérapie concomitante ou antérieure, une radiothérapie antérieure, une atteinte préexistante du parenchyme pulmonaire, des métastases pulmonaires ou des infections respiratoires. Une incidence plus élevée d’API (environ 5 % avec un taux de mortalité de 1,5 %) est observée chez les patients des études conduites au Japon.
Chez les patients qui présentent de manière inexpliquée de nouveaux symptômes pulmonaires et/ou une majoration de ces symptômes tels que dyspnée, toux et fièvre, le traitement par l’erlotinib doit être interrompu dans l'attente d’explorations diagnostiques. Les patients traités par l’erlotinib associé à la gemcitabine doivent être étroitement surveillés quant à la possibilité de développer un évènement à type d’API. En cas de diagnostic d’API, le traitement par l’erlotinib doit être arrêté et un traitement adéquat doit être instauré si nécessaire (voir rubrique 4.8).
Diarrhées, déshydratation, déséquilibre des électrolytes et insuffisance rénale
Des cas de diarrhée (dont de très rares cas fatals) sont survenus chez environ 50 % des patients traités par l’erlotinib ; les formes modérées ou sévères doivent être traitées, par exemple, par le lopéramide. Une réduction de la posologie peut parfois être nécessaire. Dans les études cliniques, les doses étaient réduites par paliers de 50 mg. Les réductions de doses par paliers de 25 mg n’ont pas été étudiées. En cas de déshydratation associée à des diarrhées, à des nausées, à une anorexie ou à des vomissements sévères et persistants, le traitement par l’erlotinib doit être interrompu et des mesures adaptées de réhydratation doivent être instaurées (voir rubrique 4.8). De rares cas d’hypokaliémie et d’insuffisance rénale (dont certains d’évolution fatale) ont été rapportés. Certains cas étaient secondaires à une déshydratation sévère due à des diarrhées, des vomissements et/ou une anorexie, alors que d’autres cas étaient liés à une chimiothérapie concomitante.
Dans les cas de diarrhées sévères ou persistantes, ou conduisant à une déshydratation, en particulier chez les patients ayant des facteurs de risques aggravants (en particulier en cas de chimiothérapie concomitante et d’autres traitements, symptômes ou pathologies ou autres facteurs prédisposants dont l’âge), le traitement par l’erlotinib doit être interrompu et des mesures appropriées de réhydratation intensive du patient par voie intraveineuse doivent être mises en oeuvre. De plus la fonction rénale et les électrolytes sériques, incluant la kaliémie, doivent être surveillés chez les patients à risque de déshydratation.
Hépatotoxicité
Perforation gastro-intestinale
Les patients recevant ERLOTINIB BIOGARAN ont un risque augmenté de perforation gastro-intestinale, qui a été peu fréquemment observée (dont certains cas ont été fatals). Les patients recevant de façon concomitante des agents anti-angiogéniques, des corticostéroïdes, des AINS, et/ou une chimiothérapie à base de taxane, ou un antécédent d’ulcère gastro-duodénal ou de diverticulose ont un risque augmenté. ERLOTINIB BIOGARAN doit être arrêté définitivement chez les patients qui développent une perforation gastrointestinale (voir rubrique 4.8).
Affections bulleuses et exfoliatives de la peau
Des cas de lésions bulleuses, phlycténulaires et exfoliatives ont été rapportés, y compris de très rares cas suggérant un syndrome de Stevens-Johnson / Syndrome de Lyell (nécrolyse épidermique toxique), qui dans certains cas ont été fatals (voir rubrique 4.8). Le traitement par ERLOTINIB BIOGARAN doit être interrompu ou arrêté définitivement si les patients présentent des lésions bulleuses ou exfoliatives sévères. Les patients présentant des lésions bulleuses et exfoliatives doivent être explorés à la recherche d’une infection cutanée et traités selon les recommandations locales.
Affections oculaires
Les patients présentant des signes et des symptômes évocateurs d'une kératite aiguë ou d’une kératite s’aggravant tels que, inflammation oculaire, larmoiement, sensibilité à la lumière, vision floue, douleur oculaire et/ou yeux rouges, doivent être adressés rapidement à un spécialiste en ophtalmologie. Si un diagnostic de kératite ulcérée est confirmé, le traitement par ERLOTINIB BIOGARAN doit être interrompu ou arrêté. Si une kératite est diagnostiquée, les bénéfices et les risques de la poursuite du traitement devront être soigneusement évalués. ERLOTINIB BIOGARAN doit être utilisé avec prudence chez les patients ayant des antécédents de kératite, kératite ulcérée ou sécheresse oculaire sévère. L'utilisation de lentilles de contact est également un facteur de risque de kératite et d’ulcération. De très rares cas de perforation ou d’ulcération de la cornée ont été rapportés lors de l’utilisation d’erlotinib (voir rubrique 4.8).
Interactions avec d’autres médicaments
Les inducteurs puissants du CYP3A4 peuvent réduire l’efficacité de l’erlotinib tandis que les inhibiteurs puissants du CYP3A4 peuvent augmenter sa toxicité. La prise concomitante de ce type de molécules doit être évitée (voir rubrique 4.5).
Autres interactions
L’erlotinib se caractérise par une diminution de solubilité à un pH supérieur à 5. Les médicaments qui modifient le pH de la partie supérieure du tractus gastro-intestinal, comme les inhibiteurs de la pompe à protons, les antagonistes H2 et les antiacides, peuvent modifier la solubilité de l’erlotinib et de ce fait sa biodisponibilité. L’augmentation de la posologie de ERLOTINIB BIOGARAN lors de sa co-administration à de tels produits ne compense probablement pas la diminution de son exposition. L’association de l’erlotinib aux inhibiteurs de la pompe à protons doit être évitée. Les effets de l’administration concomitante de l’erlotinib à des antagonistes H2 et à des antiacides ne sont pas connus ; cependant, une diminution de la biodisponibilité est probable.
Par conséquent, l’administration concomitante de ces associations doit être évitée (voir rubrique 4.5). Si l’utilisation des antiacides est jugée nécessaire durant le traitement par ERLOTINIB BIOGARAN, ils doivent être pris au moins 4 heures avant ou 2 heures après la dose quotidienne de ERLOTINIB BIOGARAN.
Ce médicament contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les études d’interactions n’ont été réalisées que chez l’adulte.
L’erlotinib et les autres substrats de CYP
In vitro l'erlotinib est un inhibiteur puissant du CYP1A1 et un inhibiteur modéré des CYP3A4 et CYP2C8, ainsi qu’un inhibiteur puissant de la glucuroconjugaison par l’UGT1A1. Du fait de la très faible expression du CYP1A1 dans les tissus humains, la pertinence physiologique d’une forte inhibition du CYP1A1 n’est pas connue.
Lors de la co-administration de l’erlotinib avec la ciprofloxacine, un inhibiteur modéré du CYP1A2, l’aire sous la courbe (ASC) de l’erlotinib a augmentée significativement de 39 % tandis qu’aucun changement significatif de la Cmax n’a été trouvé. De la même manière, l’ASC et la Cmax du métabolite actif étaient respectivement augmentées d’environ 60 % et 48 %. La pertinence clinique de cette augmentation n’a pas été établie. Une attention particulière doit être exercée lors de l’association de la ciprofloxacine ou des inhibiteurs puissants du CYP1A2 (ex : fluvoxamine) à l’erlotinib. Si des effets indésirables liés à l’erlotinib sont observés, la posologie d’erlotinib peut être diminuée.
Le prétraitement ou la co-administration d’erlotinib n’ont pas modifié la clairance des substrats spécifiques du CYP3A4, tel que le midazolam et l’érythromycine, mais semblent diminuer la biodisponibilité orale du midazolam jusqu’à 24 %. Dans une autre étude clinique, l'erlotinib n’a pas modifié les paramètres pharmacocinétiques du paclitaxel, un substrat des CYP3A4/2C8, administré concomitamment. Des interactions significatives avec la clairance d’autres substrats du CYP3A4 sont par conséquent improbables.
L’inhibition de la glucuroconjugaison pourrait entraîner des interactions avec les médicaments substrats de l’UGT1A1 et qui sont exclusivement éliminés par cette voie. Les patients avec une faible expression de l’UGT1A1 ou qui présentent des troubles génétiques de la glucuroconjugaison (ex : maladie de Gilbert) pourraient présenter une augmentation des concentrations sériques en bilirubine et devront être traités avec précaution.
Chez l'homme, l’erlotinib est métabolisé par les cytochromes hépatiques, principalement par le CYP3A4 et à un moindre degré par le CYP1A2. Le métabolisme extra hépatique par le CYP3A4 intestinal, le CYP1A1 pulmonaire et le CYP1B1 du tissu tumoral contribuent potentiellement à la clairance métabolique de l’erlotinib. Des interactions pourraient survenir avec les substances actives métabolisées par ces enzymes, ou qui les inhibent ou les induisent.
Les inhibiteurs puissants du CYP3A4 ralentissent le métabolisme de l’erlotinib et augmentent ses concentrations plasmatiques. Dans une étude clinique, l’utilisation concomitante du kétoconazole (200 mg par voie orale deux fois par jour pendant 5 jours), un inhibiteur puissant du CYP3A4, a entraîné une augmentation de 86 % de l’aire sous la courbe [ASC] et de 69 % de la Cmax de l’erlotinib. De ce fait, l’association d’erlotinib aux inhibiteurs puissants du CYP3A4, tels que les antifongiques azolés (ex : kétoconazole, itraconazole, voriconazole), les inhibiteurs de protéase, l’érythromycine ou la clarithromycine doit être faite avec prudence. Si nécessaire, la dose d’erlotinib doit être réduite, particulièrement en cas d’apparition de toxicité.
Les inducteurs puissants du CYP3A4 accélèrent le métabolisme de l’erlotinib et diminuent significativement ses concentrations plasmatiques. Dans une étude clinique, l’utilisation concomitante d’erlotinib et de rifampicine (600 mg par voie orale une fois par jour pendant 7 jours), inducteur puissant du CYP3A4, a conduit à une diminution de 69 % de la médiane de l’ASC de l’erlotinib. La co-administration de la rifampicine à une dose unique de 450 mg d’erlotinib a conduit à une moyenne de l’ASC de l’erlotinib correspondant à 57,5 % de celle obtenue avec une dose unique de 150 mg d’erlotinib en l’absence de rifampicine. Par conséquent, la co-administration d’erlotinib à des inducteurs du CYP3A4 doit être évitée.
Pour les patients nécessitant un traitement concomitant d’erlotinib avec un puissant inducteur du CYP3A4 comme la rifampicine, une augmentation de la dose jusqu’à 300 mg doit être envisagée tout en surveillant étroitement leur tolérance (notamment surveillance des fonctions rénales, hépatiques et des électrolytes sériques). Si cette dose est bien tolérée pendant plus de 2 semaines, une augmentation supplémentaire jusqu’à la dose de 450 mg pourrait être envisagée avec une surveillance étroite de la tolérance. La diminution de l’exposition à l’erlotinib pourrait également apparaître avec d’autres inducteurs tels que la phénytoïne, la carbamazépine, les barbituriques ou le millepertuis (hypericum perforatum). La prudence est de rigueur lorsque ces principes actifs sont associés à l’erlotinib. Des traitements alternatifs faiblement inducteurs du CYP3A4 doivent être envisagés chaque fois que possible.
L’erlotinib et les anticoagulants coumariniques
Des interactions avec des dérivés coumariniques, notamment la warfarine, ayant conduit à une augmentation de l’INR (International Normalized Ratio) et à des hémorragies, dans certains cas fatales, ont été rapportées chez des patients recevant de l’erlotinib. Chez les patients conjointement traités par un dérivé coumarinique, le temps de prothrombine ou l’INR doivent être régulièrement contrôlés.
L’erlotinib et les statines
L’association de l’erlotinib avec une statine peut augmenter le risque de myopathie induite par les statines (y compris rhabdomyolyse), qui a été rarement observée.
L’erlotinib et les fumeurs
Les résultats d’une étude d’interaction pharmacocinétique ont montré une diminution significative de l’aire sous la courbe (AUCinf), de la concentration plasmatique maximale (Cmax) et de la concentration plasmatique à 24 heures respectivement d’un facteur de 2,8, 1,5, et 9 après l’administration d’erlotinib chez les fumeurs par rapport aux non-fumeurs. Par conséquent, les patients continuant à fumer devront être encouragés à arrêter le plus tôt possible avant le début du traitement par ERLOTINIB BIOGARAN compte-tenu de la réduction des concentrations plasmatiques d’erlotinib. En se basant sur les données issues de l’étude CURRENTS, aucune preuve n’a mis en évidence un quelconque bénéfice de la posologie la plus élevée de 300 mg d’erlotinib par rapport à la posologie recommandée de 150 mg chez les fumeurs. Les données de tolérance étaient comparables entre la posologie de 300 mg et celle de 150 mg. Cependant, il y a eu une augmentation de l’incidence des éruptions cutanées, des affections pulmonaires interstitielles et des diarrhées, chez les patients recevant la posologie la plus élevée d’erlotinib (voir les rubriques 4.2, 4.4, 5.1 et 5.2).
L’erlotinib et les inhibiteurs de la glycoprotéine-P
L’erlotinib est un substrat de la glycoprotéine-P. L’administration concomitante des inhibiteurs de la glycoprotéine-P tels que la ciclosporine et le vérapamil, peut conduire à une altération de la distribution et/ou de l’élimination de l’erlotinib. Les conséquences de cette interaction, par exemple au niveau de la toxicité pour le SNC, n’ont pas été établies. Une attention particulière doit être exercée dans de telles situations.
L’erlotinib et les médicaments qui modifient le pH
L’erlotinib se caractérise par une diminution de solubilité à un pH supérieur à 5. Les médicaments qui modifient le pH de la partie supérieure du tractus gastro-intestinal peuvent modifier la solubilité de l’erlotinib et de ce fait sa biodisponibilité. La co-administration de l’erlotinib avec l’oméprazole, un inhibiteur de la pompe à protons (IPP), a diminué l’aire sous la courbe (ASC) et la concentration maximale (Cmax) de l’erlotinib respectivement de 46 % et 61 %. Il n’y avait pas de modification du Tmax ou de la demi-vie. L’administration concomitante d’erlotinib à 300 mg de ranitidine, un antagoniste du récepteur H2, a diminué l’aire sous la courbe (ASC) et la concentration maximale (Cmax) de l’erlotinib respectivement de 33 % et 54 %. L’augmentation de la posologie de l’erlotinib lors de sa co-administration à de tels produits, ne compense probablement pas la diminution de son exposition. Cependant, lorsque l’erlotinib administré de façon espacée, 2 heures avant ou 10 heures après l’administration de ranitidine 150 mg deux fois par jour a été dosé, l’aire sous la courbe (ASC) et la concentration maximale (Cmax) de l’erlotinib ont seulement diminués respectivement de 15 % et 17 %. L’effet des antiacides sur l’absorption de l’erlotinib n’a pas été étudié, mais l’absorption peut être altérée, conduisant à une diminution des taux plasmatiques. En résumé, l’association de l’erlotinib aux inhibiteurs de la pompe à protons doit être évitée. Si l’utilisation des antiacides est jugée nécessaire durant le traitement par ERLOTINIB BIOGARAN, ils doivent être pris au moins 4 heures avant ou 2 heures après la dose quotidienne de ERLOTINIB BIOGARAN. Si l’utilisation de la ranitidine est envisagée, elle doit l’être de façon espacée ; par exemple ERLOTINIB BIOGARAN doit être pris au moins 2 heures avant ou 10 heures après la ranitidine.
L’erlotinib et la gemcitabine
Dans une étude de phase Ib, il n’y a eu aucun effet significatif de la gemcitabine sur les paramètres pharmacocinétiques de l’erlotinib ni de l’erlotinib sur ceux de la gemcitabine.
L’erlotinib et le carboplatine/paclitaxel
L’erlotinib augmente les concentrations en sel de platine. Dans une étude clinique, l’utilisation concomitante de l’erlotinib au carboplatine et au paclitaxel a conduit à une augmentation de 10,6 % de l’ASC0-48 du sel de platine total. Bien que statistiquement significative, l’importance de cette différence n’est pas considérée comme cliniquement pertinente.
En pratique clinique, d’autres facteurs associés peuvent conduire à une augmentation de l’exposition au carboplatine comme une altération de la fonction rénale. Il n’y a pas eu d’effets significatifs du carboplatine ou du paclitaxel sur les paramètres pharmacocinétiques de l’erlotinib.
L’erlotinib et la capécitabine
La capécitabine peut augmenter les concentrations de l’erlotinib. Lorsque l’erlotinib a été associé à la capécitabine, il y a eu une augmentation significative de l’ASC de l’erlotinib et une augmentation limitée de la Cmax par rapport aux valeurs observées dans une autre étude dans laquelle l’erlotinib a été administré seul. Il n’y a pas eu d’effets significatifs de l’erlotinib sur les paramètres pharmacocinétiques de la capécitabine.
L'erlotinib et les inhibiteurs du protéasome
Compte tenu de leur mécanisme d’action, les inhibiteurs du protéasome, y compris le bortezomib, pourraient avoir une influence sur l'effet des inhibiteurs de l'EGFR, notamment l'erlotinib. Cette influence est étayée par des données cliniques limitées et des études précliniques montrant une dégradation de l’EGFR par le protéasome.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n’existe pas de données suffisantes relatives à l’utilisation d’erlotinib chez la femme enceinte. Les études réalisées chez l’animal n’ont pas mis en évidence de tératogénicité ou de parturition anormale. Cependant, un effet indésirable sur la grossesse ne peut être exclu car des études réalisées chez le rat et le lapin ont montré une létalité embryo-fœtale augmentée (voir rubrique 5.3). Le risque potentiel chez l'homme est inconnu.
Femmes en âge de procréer
Les femmes en âge de procréer doivent être incitées à éviter une grossesse pendant le traitement par ERLOTINIB BIOGARAN. Une méthode de contraception efficace doit être utilisée pendant le traitement et pendant au moins les 2 semaines qui suivent la fin de celui-ci. En cas de survenue d’une grossesse, le traitement ne doit être poursuivi que si le bénéfice attendu pour la mère justifie le risque pris pour le fœtus.
Il n’existe pas de données sur l’excrétion de l’erlotinib dans le lait maternel. Aucune étude n’a été menée pour évaluer l’impact de l’erlotinib sur la production de lait ou sa présence dans le lait maternel. En raison du potentiel délétère inconnu pour le nourrisson, l’allaitement est déconseillé lors d’un traitement par erlotinib et pendant au moins les 2 semaines suivant la dernière dose.
Fertilité
Les études réalisées chez l’animal n’ont pas mis en évidence de trouble de la fécondité. Cependant, un effet indésirable sur la fécondité ne peut être exclu car les études réalisées chez l’animal ont montré des effets sur les paramètres de la reproduction (voir rubrique 5.3). Le risque potentiel chez l'homme est inconnu.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de tolérance
L’évaluation de la tolérance de l’erlotinib est basée sur des données issues de plus de 1500 patients traités par au moins une dose de 150 mg d’erlotinib en monothérapie, et plus de 300 patients ayant reçu de l’erlotinib 100 mg ou 150 mg en association avec gemcitabine.
Cancer bronchique non à petites cellules (Erlotinib en monothérapie)
Première ligne de traitement chez des patients présentant des mutations de l’EGFR
Dans une étude de phase III en ouvert, randomisée, ML20650, conduite chez 154 patients, la tolérance de l’erlotinib en première ligne de traitement des patients atteints d’un CBNPC présentant des mutations activatrices de l’EGFR a été évaluée chez 75 patients.
Les effets indésirables les plus fréquemment observés chez les patients traités par erlotinib dans l’étude ML20650 étaient une éruption cutanée et une diarrhée, la plupart étaient de sévérité de grade 1/2 et gérables sans intervention. Des informations complètes sur le grade et l’incidence des éruptions cutanées et des diarrhées pour toutes les études cliniques sont disponibles dans la rubrique « Description d’effets indésirables sélectionnés » ci-dessous.
Traitement de maintenance
Dans deux autres études de phase III en double aveugle, randomisées, contrôlées par placebo, BO18192 (SATURN) et BO25460 (IUNO), l’erlotinib a été administré en maintenance après une première ligne de chimiothérapie. Ces études ont été conduites chez 1532 patients atteints d’un CBNPC avancé, récurrent ou métastatique, à la suite d’une chimiothérapie standard de première ligne à base de sels de platine.
Les effets indésirables les plus fréquemment observés chez les patients traités par erlotinib dans les études BO18192 et BO25460 étaient une éruption cutanée et une diarhée.
Deuxième ligne de traitement et plus
Dans une étude randomisée en double aveugle (BR.21 : erlotinib administré en deuxième ligne de traitement), les effets indésirables (EI) les plus fréquemment observés ont été des éruptions cutanées et des diarrhées. La plupart ont été de grade 1/2 et n’ont pas nécessité d’intervention spécifique. Le délai médian de survenue des éruptions cutanées a été de 8 jours et celui des diarrhées de 12 jours.
Cancer du pancréas (erlotinib associé à la gemcitabine)
Les effets indésirables les plus fréquents dans l’étude pivot PA.3 chez des patients atteints d’un cancer du pancréas recevant de l’erlotinib 100 mg associé à la gemcitabine étaient une fatigue, une éruption cutanée et une diarrhée. Le délai médian de survenue d’une éruption cutanée et d’une diarrhée était de 10 jours et 15 jours, respectivement.
Liste tabulée des effets indésirables
L’incidence des effets indésirables (EI) observés, lors des essais cliniques et depuis la commercialisation, avec l’erlotinib seul ou en association avec une chimiothérapie est résumée dans le Tableau 1. Les effets indésirables sont listés par classe de systèmes d’organes (MedDRA). La catégorie de fréquence correspondant à chaque effet indésirable est basée sur la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
|
Infections et infestations |
|
|||
|
Très fréquent |
infection * |
|
||
|
Troubles du métabolisme et de la nutrition |
|
|||
|
Très fréquent |
anorexie, diminution du poids |
|
||
|
Affections psychiatriques |
|
|||
|
Très fréquent |
dépression |
|
||
|
Affections du système nerveux |
|
|||
|
Très fréquent |
neuropathie, maux de tête |
|
||
|
Affections oculaires |
|
|||
|
Très fréquent |
kératoconjontivites sèches |
|
||
|
Fréquent |
kératites, conjontivites |
|
||
|
Peu fréquent |
modification des cils * |
|
||
|
Très rare |
perforation de la cornée, ulcération de la cornée, uvéites |
|
||
|
Affections respiratoires, thoraciques et médiastinales |
|
|||
|
Très fréquent |
dyspnée, toux |
|
||
|
Fréquent |
epistaxis |
|
||
|
Peu fréquent |
Pneumopathie interstitielle diffuse * |
|
||
|
Affections gastrointestinales |
|
|||
|
Très fréquent |
diarrhée *, nausée, vomissements, stomatites, douleurs abdominales, dyspepsie, flatulence |
|
||
|
Fréquent |
hémorragies gastro-intestinales * |
|
||
|
Peu fréquent |
perforations gastro-intestinales * |
|
||
|
Rare |
pneumatose intestinale |
|
||
|
Affections hépatobiliaires |
|
|||
|
Très fréquent |
anomalies des explorations fonctionnelles hépatiques * |
|
||
|
Rare |
insuffisance hépatique *, hépatite |
|
||
|
Fréquence indéterminée (ne peut être estimée selon les données disponibles) |
hépatite aiguë |
|
||
|
Affections de la peau et du tissu sous-cutané |
|||
|
Très fréquent |
éruptions cutanées *, prurit |
||
|
Fréquent |
alopécie, sécheresse cutanée, paronychie, folliculite, acné/dermatite acnéiforme, fissures de la peau |
||
|
Peu fréquent |
hirsutisme, modification des sourcils, ongles cassants et perte des ongles, réactions cutanées légères telles que hyperpigmentation |
||
|
Rare |
syndrome d’érythrodys-esthésie palmo-plantaire |
||
|
Très rare |
syndrome de Stevens-Johnson / nécrolyse épidermique toxique * |
||
|
Affections du rein et des voies urinaires |
|||
|
Fréquent |
insuffisance rénale |
||
|
Peu fréquent |
néphrite, protéinurie |
||
|
Troubles généraux et anomalies au site d’administration |
|||
|
Très fréquent |
asthénie, fièvre, frissons |
||
Description d’effets indésirables sélectionnés
Eruption cutanée
L’éruption cutanée inclut la dermatite acnéiforme. En général, l’éruption cutanée se manifeste comme une éruption érythémateuse et papulopustuleuse légère ou modérée, qui peut survenir ou s’aggraver dans les zones exposées au soleil. Pour les patients qui s’exposent au soleil, des vêtements protecteurs, et/ou l’usage d’écran solaire (ex. : filtre minéral) peuvent être conseillés.
Diarrhée
La diarrhée peut entraîner une déshydratation, une hypokaliémie et une insuffisance rénale. Cela peut conduire à des décès (voir rubrique 4.4).
Tableau 2 : Résumé de l’incidence et du grade d’éruption cutanée et de diarrhée observés dans chaque étude clinique
|
Indication |
Eruption cutanée (%) |
Diarrhée (%) |
|||||||||
|
Grade |
Action prise |
Grade |
Action prise |
||||||||
|
Tout |
3 |
4 |
Arrêt |
Mod1 |
Tout |
3 |
4 |
Arrêt |
Mod1 |
||
|
ML20650 |
CBNPC |
80 |
9 |
0 |
1 |
11 |
57 |
4 |
0 |
1 |
7 |
|
BO18192 |
CBNPC |
49,2 |
6,0 |
0 |
1 |
8.3 |
20,3 |
1.8 |
0 |
<1 |
3 |
|
BO25460 |
CBNPC |
39,4 |
5,0 |
0 |
0 |
5.6 |
24,2 |
2,5 |
0 |
0 |
2,8 |
|
BR.21 |
CBNPC |
75 |
9 |
1 |
6 |
54 |
6 |
1 |
1 |
||
|
PA.3 |
Cancer du pancréas |
- |
5 |
1 |
2 |
- |
5 |
1 |
2 |
||
Infection
Pneumopathie interstitielle diffuse
Des pneumopathies interstitielles diffuses, parfois d’issue fatale, ont été observées chez des patients recevant erlotinib pour le traitement de CBNPC ou d’autres tumeurs solides avancées (voir rubrique 4.4). Une incidence plus élevée a été observée chez les patients au Japon (voir rubrique 4.4).
Hémorragies gastro-intestinales
Les hémorragies gastro-intestinales peuvent être fatales (voir rubrique 4.4). Au cours des études cliniques, certains cas ont été associés à une administration concomitante de warfarine et d’autres à une administration concomitante d’AINS (voir rubrique 4.5). Les perforations gastro-intestinales peuvent également être fatales (voir rubrique 4.4).
Anomalies des explorations fonctionnelles hépatiques
Les anomalies comprennent une augmentation de l’alanine aminotransférase [ALAT], de l’aspartate aminotransférase [ASAT] et de la bilirubine. Les cas étaient principalement de sévérité légère à modérée, transitoires ou associés à des métastases hépatiques.
Insuffisance hépatique
Cela inclut des cas de décès. Les facteurs de risque peuvent inclure une maladie hépatique préexistante ou la prise concomitante de médicaments hépatotoxiques (voir rubrique 4.4).
Syndrome de Stevens-Johnson/Nécrolyse épidermique toxique
Cela inclut des cas de décès (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Symptômes
Des doses uniques d’erlotinib par voie orale allant jusqu’à 1 000 mg d’erlotinib chez des volontaires sains et jusqu’à 1 600 mg chez des patients atteints d’un cancer ont été bien tolérées. L’administration d’une dose de 200 mg deux fois par jour a été mal tolérée par des volontaires sains au bout de seulement quelques jours de traitement. Les données issues de ces études indiquent que des effets indésirables sévères tels que diarrhées, éruptions cutanées et, possiblement augmentation de l’activité des aminotransférases hépatiques pourraient survenir au-delà de la dose recommandée.
Prise en charge d’un surdosage
En cas de suspicion de surdosage, l'administration de ERLOTINIB BIOGARAN doit être suspendue et un traitement symptomatique doit être instauré.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
L’erlotinib est un inhibiteur de la tyrosine kinase du récepteur du facteur de croissance épidermique humain de type 1 (Epidermal Growth Factor Receptor (EGFR) également connu comme HER1). L’erlotinib est un puissant inhibiteur de la phosphorylation intracellulaire de l’EGFR. L’EGFR est exprimé à la surface de cellules normales et cancéreuses. Dans des modèles non cliniques, l'inhibition de la phosphotyrosine de l’EGFR résulte en un arrêt de la prolifération et/ou à une mort cellulaire. Des mutations de l’EGFR peuvent conduire à une activation constitutive des voies de signalisation anti-apoptotique et de la prolifération. La puissante efficacité de l'erlotinib sur le blocage de la signalisation médiée par EGFR dans ces tumeurs arborant des mutations positives de l'EGFR est attribuée à la liaison étroite de l'erlotinib au site de liaison de l’ATP dans le domaine de la kinase mutée de l'EGFR. En raison du blocage en aval de la signalisation, la prolifération des cellules est arrêtée, et la mort cellulaire est induite par la voie intrinsèque de l'apoptose. La régression de la tumeur est observée dans des modèles de souris où l’expression de ces mutations activatrices de l’EGFR est renforcée.
Efficacité clinique
Traitement en première ligne du cancer bronchique non à petites cellules (CBNPC) chez des patients présentant des mutations activatrices de l’EGFR (Erlotinib en monothérapie) :
L’efficacité de l’erlotinib en première ligne de traitement des patients ayant un CBNPC présentant des mutations activatrices de l’EGFR a été démontrée dans un essai de phase III, randomisé, en ouvert (ML20650, EURTAC). Cette étude a été conduite chez des patients caucasiens atteints d’un CBNPC localement avancé ou métastatique (stades IIIB et IV) qui n’avaient reçu précédemment ni chimiothérapie ni traitement anticancéreux systémique pour leur maladie localement avancée et qui présentaient des mutations dans le domaine tyrosine kinase de l’EGFR (délétion de l’exon 19 ou mutation de l’exon 21). Les patients ont été affectés par randomisation 1 :1 à un traitement par l’erlotinib 150 mg 1 fois par jour ou jusqu’à 4 cycles de chimiothérapie à base de doublet de sel de platine. Le critère principal était la survie sans progression (Progression Free Survival : PFS) évaluée par l’investigateur. Les résultats d'efficacité sont résumés dans le tableau 3.
Figure 1 : Courbe de Kaplan-Meier de la PFS évaluée par l’investigateur dans l’essai ML20650
(EURTAC) (cut-off d’avril 2012)
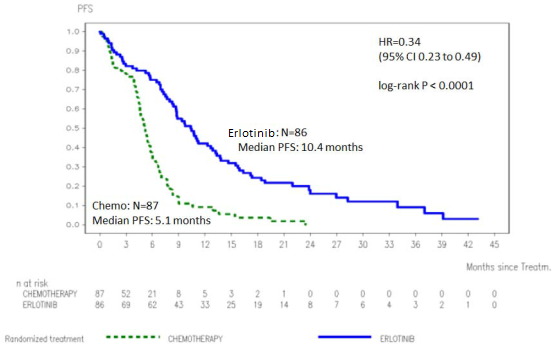
Tableau 3 : Résultats d’efficacité de l’erlotinib versus chimiothérapie de l’essai ML20650 (EURTAC)
|
|
|
Erlotinib |
Chimio- thérapie |
Hazard Ratio (IC 95%) |
Valeur du p |
|
Analyse intermédiaire planifiée (OS à 35 % de maturité)
(n=153) Date de Cut-off : Août 2010 |
|
n=77 |
n=76 |
|
|
|
|
9,4 |
5,2 |
0,42 |
p<0,0001 |
|
|
|
|||||
|
Critère d’évaluation primaire : Survie sans progression (PFS, médiane en mois) * Evaluée par l’investigateur** |
|||||
|
|
|
|
[0,27-0,64] |
|
|
|
Revue indépendante** |
10,4 |
5,4 |
0,47 |
p=0,003 |
|
|
[0,27-0,78] |
|||||
|
Meilleur taux de réponse globale (RC/RP) |
54,5% |
10,5% |
|
p<0,0001 |
|
|
(OS) (mois) |
22,9 |
18,8 |
0,80 [0,47-1,37] |
p=0,4170 |
|
|
Analyse exploratoire (OS à 40 % de maturité) (n=173)
Cut-off date: Janvier 2011 |
|
n=86 |
n=87 |
|
|
|
PFS (médiane en mois), Evaluée par l’investigateur |
9,7 |
5,2 |
0,37 [0,27-0,54] |
p<0,0001 |
|
|
Meilleur taux de réponse globale (RC/RP) |
58,1% |
14,9% |
|
p<0,0001 |
|
|
OS (mois) |
19,3 |
19,5 |
1,04 [0,65-1,68] |
p=0,8702 |
|
|
Analyse actualisée
(OS à 62 % de maturité) (n=173)
Cut-off date: Avril 2012 |
|
n=86 |
n=87 |
|
|
|
PFS (médiane en mois) |
10,4 |
5,1 |
0,34 [0,23-0,49] |
p<0,0001 |
|
|
OS*** (mois) |
22,9 |
20,8 |
0,93 [0,64-1,36] |
p=0,7149 |
RC= réponse complète ; RP = réponse partielle
* Une diminution de 58 % du risque de progression de la maladie ou de décès a été observée.
** Le taux de concordance entre l’évaluation de l’investigateur et celle du comité de revue indépendant était de 70 %.
*** Un taux élevé de cross-over a été observé avec 82 % des patients du bras traité par chimiothérapie ayant reçu ultérieurement un inhibiteur de la tyrosine kinase de l’EGFR, et tous ces patients excepté deux ayant reçu l’erlotinib.
Traitement de maintenance du CBNPC après une première ligne de chimiothérapie (Erlotinib en monothérapie) :
L’efficacité et la tolérance de l’erlotinib dans le traitement de maintenance du CBNPC après une première ligne de chimiothérapie ont été étudiées dans un essai randomisé en double aveugle contrôlé versus placebo (B018192, SATURN). Cette étude a été conduite chez 889 patients atteints d’un CBNPC localement avancé ou métastatique sans progression de la maladie après 4 cycles de chimiothérapie à base de doublet de sel de platine. Les patients ont été affectés par randomisation 1:1 à un traitement par l’erlotinib 150 mg ou placebo, par voie orale, une fois par jour, jusqu’à progression de la maladie. Le critère principal de l’étude était la survie sans progression (Progression Free Survival : PFS) chez tous les patients. Les caractéristiques démographiques et pathologiques à l’inclusion des patients étaient bien équilibrées entre les deux bras de traitement. Les patients ayant un indice de performance ECOG PS>1, des co-morbidités hépatiques ou rénales significatives, n’étaient pas inclus dans l’étude.
Dans cette étude, l’ensemble de la population a montré un bénéfice pour le critère d’évaluation principal qui était la PFS (risque relatif (Hazard Ratio : HR) =0,71 p<0,0001) et pour le critère d’évaluation secondaire qui était la survie globale (overall survival : OS) (HR=0,81 p=0,0088). Cependant, le plus large bénéfice a été observé dans une analyse exploratoire prédéfinie chez des patients avec mutations activatrices de l’EGFR (n=49) en traduisant un bénéfice substantiel pour la PFS (HR=0,10 ; IC 95% ; 0,04 à 0,25 ; p<0,0001) et pour la survie globale avec un HR qui était de 0,83 (IC 95% ; 0,34 à 2,02). 67 % des patients du sous-groupe placebo avec la mutation EGFR positive ont reçu en seconde (ou plus tardive) ligne de traitement EGFR-TKIs.
L’étude BO25460 (IUNO) a été menée chez 643 patients atteints d’un CBNPC avancé sans mutation activatrice de l’EGFR de la tumeur (délétion de l’exon 19 ou mutation L858R de l’exon 21) et sans progression de la maladie après 4 cycles de chimiothérapie à base de platine.
L’objectif de l’étude était de comparer la survie globale d’une thérapie par l’erlotinib en première ligne de traitement de maintenance versus erlotinib administré au moment de la progression de la maladie. L’étude n’a pas atteint son critère d’évaluation principal. La survie globale de l’erlotinib en première ligne de traitement de maintenance n’était pas supérieure au traitement de l’erlotinib en seconde ligne de traitement chez les patients sans mutation activatrice de l’EGFR de la tumeur (HR=1,02 ; IC 95% ; 0,85 à 1,22 ; p=0,82). Le critère d’évaluation secondaire PFS n’a pas montré de différence entre erlotinib et placebo en traitement de maintenance (HR=0,94 ; IC 95% ; 0,80 à 1,11 ; p=0,48). Sur la base des données de l’étude BO25460 (IUNO), l’utilisation de l’erlotinib n’est pas recommandée en première ligne de traitement de maintenance chez les patients sans mutation activatrice de l’EGFR.
Traitement du CBNPC après échec d’au moins un régime de chimiothérapie (Erlotinib en monothérapie) :
L’efficacité et la tolérance de l’erlotinib en traitement de deuxième/troisième ligne ont été démontrées dans un essai randomisé en double aveugle contrôlé versus placebo (BR.21) chez 731 patients atteints d’un CBNPC localement avancé ou métastatique après échec d'au moins une ligne de chimiothérapie.
Les patients ont été affectés par randomisation 2 :1 à un traitement par l’erlotinib 150 mg ou placebo, par voie orale, une fois par jour. Les critères d’évaluation de l’étude étaient notamment la survie globale, la survie sans progression (Progression Free Survival : PFS), le taux et la durée de réponse, le délai d’aggravation des symptômes liés au cancer du poumon (toux, dyspnée et douleurs), et la tolérance. Le critère principal de l’étude était la survie.
Les caractéristiques démographiques étaient bien équilibrées entre les deux groupes de traitement. Environ deux tiers des patients étaient de sexe masculin et l'indice de performance initial (Eastern Cooperative Oncology Group ECOG - performance status (PS)) était de 2 chez environ un tiers des patients et de 3 chez 9 % des patients. Une chimiothérapie incluant un sel de platine avait été antérieurement administrée chez 93 % des patients du groupe erlotinib et chez 92 % des patients du groupe placebo, et respectivement 36 % et 37 % des patients avaient été traités par un taxane.
Le Risque Relatif (Hazard Ratio (HR)) ajusté de décès dans le groupe erlotinib par rapport au groupe placebo a été de 0,73 (IC95% : 0,60 à 0,87) (p=0,001). Le pourcentage de patients en vie à 12 mois a été de 31,2 % dans le groupe erlotinib et de 21,5 % dans le groupe placebo. La médiane de survie globale était de 6,7 mois dans le groupe erlotinib (IC95% : 5,5 à 7,8 mois) comparée à 4,7 mois dans le groupe placebo (IC95% : 4,1 à 6,3 mois).
L’effet sur la survie globale était exploré à travers différents sous-groupes de patients. Les effets de l’erlotinib sur la survie globale étaient similaires chez les patients dont l’ECOG – PS initial était de 2-3 (HR = 0,77 ; IC95%: 0,6-1,0) ou de 0-1 (HR = 0,73 ; IC95%: 0,6-0,9), les hommes (HR = 0,76 ; IC95%: 0,6-0,9) ou les femmes (HR = 0,80 ; IC95%: 0,6-1,1), les patients âgés de moins de 65 ans (HR = 0,75 ; IC95%: 0,6-0,9) ou les patients plus âgés (HR = 0,79 ; IC95%: 0,6-1,0), les patients ayant reçu auparavant un seul traitement de chimiothérapie (HR = 0,76, IC95%: 0,6-1,0), ou plus de un traitement de chimiothérapie (HR = 0,75 ; IC95%: 0,6-1,0), les patients Caucasiens (HR=0,79 ; IC95%: 0,6-1,0) ou Asiatiques (HR = 0,61 ; IC95%: 0,4-1,0), les patients avec un adénocarcinome (HR= 0,71 ; IC95%: 0,6-0,9) ou un carcinome épidermoïde (HR = 0,67 ; IC95%: 0,5-0,9), mais pas chez les patients avec d’autres types histologiques (HR 1,04 ; IC95%: 0,7-1,5), les patients diagnostiqués au stade IV (HR = 0,92 ; IC95%: 0,7-1,2) ou diagnostiqué à un stade < IV (HR = 0,65 ; IC95%: 0,5-0,8). Le bénéfice de l’erlotinib a été meilleur chez les patients n’ayant jamais fumé (HR survie : 0,42 ; IC95% : 0,28-0,64) comparé aux fumeurs ou anciens fumeurs (HR = 0,87 ; IC95% : 0,71- 1,05).
Parmi les 45 % de patients dont le statut d’expression EGFR était connu, le Hazard Ratio était de 0,68 (IC95%: 0,49-0,94) pour les patients avec des tumeurs EGFR-positif et de 0,93 (IC95%: 0,63-1,36) pour les patients avec des tumeurs EGFR-négatif (déterminé par IHC en utilisant le kit EGFR pharmaDx et définissant le statut EGFR-négatif comme taux inférieur à 10% des cellules tumorales colorées).Chez les 55% de patients restants, dont le statut d’expression EGFR était inconnu, le HR était de 0,77 (IC95%: 0,61- 0,98).
La survie médiane sans progression (PFS) était de 9,7 semaines dans le groupe erlotinib (IC95% : 8,4 à 12,4 semaines) comparée à 8,0 semaines dans le groupe placebo (IC 95% : 7,9 à 8,1 semaines).
Le taux de réponse objective selon les critères RECIST (Response Evaluation Criteria in Solid Tumors) a été de 8,9 % (IC95% : 6,4 à 12,0 %) dans le groupe erlotinib. Les 330 premiers patients ont été évalués de manière centralisée (taux de réponse : 6,2 %) ; 401 patients ont été évalués par les investigateurs (taux de réponse : 11,2 %). La durée médiane de réponse a été de 34,3 semaines (allant de 9,7 à plus de 57,6 semaines). La proportion des patients ayant présenté une réponse complète ou partielle ou une stabilisation de la maladie a été de 44,0 % dans le groupe erlotinib et de 27,5 % dans le groupe placebo (p=0,004).
Un bénéfice en survie a été également observé chez les patients traités par l’erlotinib n'ayant pas présenté une réponse tumorale objective (selon les critères RECIST). Cela a été montré avec un HR de décès de 0,82 (IC95% : 0,68 à 0,99) chez les patients dont la meilleure réponse a été une stabilisation ou une progression de la maladie. L’erlotinib a exercé un effet bénéfique en prolongeant significativement les délais d’aggravation de la toux, de la dyspnée et des douleurs comparativement au placebo.
Dans une étude de phase III en double aveugle, randomisée (MO22162, CURRENTS) comparant deux posologie d’erlotinib (300 mg vs. 150 mg) chez des fumeurs (moyenne de 38 paquets par an) atteints d’un CBNPC localement avancé ou métastatique en seconde ligne de traitement après échec d’une chimiothérapie, la posologie de 300 mg d’erlotinib n’a pas démontré de bénéfice en PFS par rapport à la posologie recommandée (7,00 vs. 6,86 semaines, respectivement).
Les critères secondaires d’efficacité étaient tous cohérents avec le critère principal et aucune différence de survie globale n’a été relevée entre les patients traités par erlotinib à la posologie de 300 mg et de 150 mg par jour (HR 1,03; IC 95%; 0,8 à 1,32). Les données de tolérance étaient comparables entre la posologie de 300 mg et de 150 mg. Cependant, il y a eu une augmentation de l’incidence des éruptions cutanées, des affections pulmonaires interstitielles et des diarrhées, chez les patients recevant la posologie la plus élevée d’erlotinib. En se basant sur les données issues de l’étude CURRENTS, aucune preuve n’a mis en évidence un quelconque bénéfice de la posologie la plus élevée de 300 mg d’erlotinib par rapport à la posologie recommandée de 150 mg chez les fumeurs.
Les patients de cette étude n’étaient pas sélectionnés selon le statut des mutations de l’EGFR (voir les rubriques 4.2, 4.4, 4.5 et 5.2).
Cancer du pancréas (Erlotinib associé à la gemcitabine dans l’étude PA.3):
L’efficacité et la tolérance de l’erlotinib associé à la gemcitabine en traitement de première ligne ont été évaluées dans un essai randomisé, en double aveugle, contrôlé versus placebo chez des patients atteints d'un cancer du pancréas localement avancé, non résécable ou métastatique. Les patients ont été randomisés pour recevoir un traitement par l’erlotinib ou placebo une fois par jour en traitement continu plus gemcitabine IV (1000 mg/m², Cycle 1 - jours 1, 8, 15, 22, 29, 36 et 43 d’un cycle de 8 semaines ; cycle 2 et cycles ultérieurs - jours 1, 8 et 15 d’un cycle de 4 semaines [posologie et rythme d’administration de la gemcitabine approuvés dans le traitement du cancer du pancréas : voir le RCP de la gemcitabine]). L’erlotinib ou le placebo ont été pris par voie orale une fois par jour jusqu'à progression de la maladie ou apparition d’une toxicité inacceptable. Le critère principal de l’étude était la survie globale.
Les caractéristiques démographiques et pathologiques à l’inclusion des patients étaient similaires entre les deux groupes de traitement, erlotinib 100 mg plus gemcitabine ou placebo plus gemcitabine, à l’exception d’une proportion légèrement plus élevée de femmes dans le groupe erlotinib/gemcitabine que dans le groupe placebo/gemcitabine:
|
A l’inclusion |
Erlotinib |
Placebo |
|
Femmes |
51 % |
44 % |
|
Indice de performance ECOG (PS) = 0 |
31 % |
32 % |
|
Indice de performance ECOG (PS) = 1 |
51 % |
51 % |
|
Indice de performance ECOG (PS) = 2 |
17 % |
17 % |
|
Maladie métastatique à l’inclusion |
77 % |
76 % |
La survie a été évaluée dans la population en intention de traiter sur la base des données obtenues lors du suivi de la survie. Les résultats sont présentés dans le tableau ci-dessous (les résultats du groupe de patients métastatique et localement avancé proviennent d’une analyse exploratoire des sous-groupes)
|
Résultats |
Erlotinib (mois) |
Placebo (mois) |
Δ (mois) |
IC du Δ |
HR |
IC du HR |
P- value |
|
Population globale |
|||||||
|
Médiane de survie globale |
6,4 |
6,0 |
0,41 |
-0,54-1,64 |
0,82 |
0,69-0,98 |
0,028 |
|
Moyenne de survie globale |
8,8 |
7,6 |
1,16 |
-0,05-2,34 |
|||
|
Population métastatique |
|||||||
|
Médiane de survie globale |
5,9 |
5,1 |
0,87 |
-0,26-1,56 |
0,80 |
0,66-0,98 |
0,029 |
|
Moyenne de survie globale |
8,1 |
6,7 |
1,43 |
0,17-2,66 |
|||
|
Population localement avancé |
|||||||
|
Médiane de survie globale |
8,5 |
8,2 |
0,36 |
-2,43-2,96 |
0,93 |
0,65-1,35 |
0,713 |
|
Moyenne de survie globale |
10,7 |
10,5 |
0,19 |
2,43-2,69 |
|||
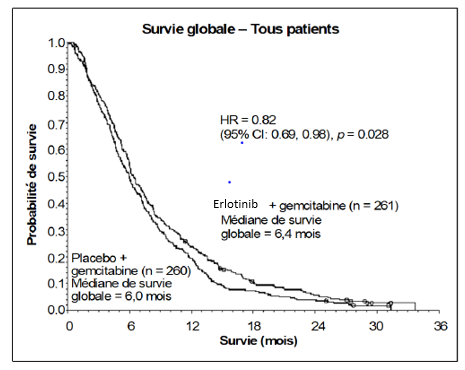
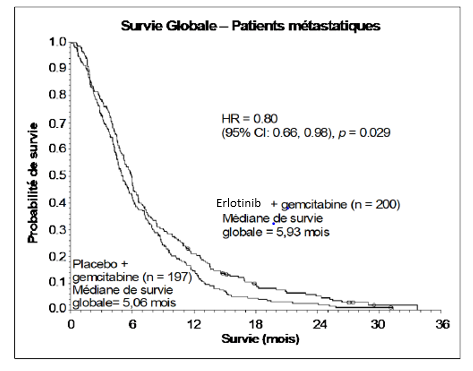
Dans une analyse post-hoc, les patients ayant un état clinique favorable à l’inclusion (faible intensité de douleur, bonne qualité de vie et bon indice de performance), peuvent tirer un meilleur bénéfice d’erlotinib. Le bénéfice est principalement lié à la présence de douleur de faible intensité.
Dans une analyse post-hoc, les patients sous erlotinib ayant développé une éruption cutanée avaient une survie globale plus longue que les patients n’ayant pas développé d’éruption cutanée (médiane de survie globale 7,2 mois contre 5 mois, risque relatif HR : 0,61).
90% des patients sous erlotinib ont développé une éruption cutanée dans les 44 premiers jours. Le temps médian d’apparition de l’éruption cutanée était de 10 jours.
Population pédiatrique
L’Agence européenne du médicament a accordé une dérogation à l’obligation de soumettre les résultats des études réalisées avec de l’erlotinib dans tous les sous-groupes de la population pédiatrique dans les indications du cancer bronchique non à petites cellules et du cancer du pancréas (voir rubrique 4.2 pour information sur l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
Après administration orale, le pic de concentration plasmatique d’erlotinib est obtenu après environ 4 heures. La biodisponibilité absolue a été estimée à 59% dans une étude chez des volontaires sains.
La prise d’aliments peut augmenter l’exposition après une prise orale.
Distribution
La valeur moyenne du volume apparent de distribution de l’erlotinib est de 232 litres. L’erlotinib diffuse dans les tissus tumoraux chez l’Homme. Lors d’une étude menée chez 4 patients, dont 3 atteints d’un cancer bronchique non à petites cellules (CBNPC) et 1 d'un cancer du larynx, recevant une dose orale quotidienne de 150 mg d’erlotinib, des dosages effectués sur des prélèvements tumoraux obtenus par excision chirurgicale au 9 ème jour de traitement ont indiqué des concentrations intra tumorales moyennes d’erlotinib de 1,185 ng/g de tissu, ce qui correspond en moyenne à 63% (intervalle: 5 – 161%) des concentrations plasmatiques maximales observées à l’état d’équilibre. Les principaux métabolites actifs étaient présents dans la tumeur à une concentration moyenne de 160 ng/g de tissu, soit globalement en moyenne 113% (intervalle : 88 – 130%) des concentrations plasmatiques maximales déterminées à l’état d’équilibre. La liaison aux protéines plasmatiques est d’environ 95%. L’erlotinib se lie à l’albumine sérique et à l’alpha-1 glycoprotéine acide (α1GPA).
Biotransformation
L’erlotinib est métabolisé par les cytochromes hépatiques chez l'homme, principalement par le CYP3A4 et, à un moindre degré, par le CYP1A2. Le métabolisme extra hépatique par le CYP3A4 intestinal, le CYP1A1 pulmonaire et le CYP1B1 du tissu tumoral contribuent potentiellement à la clairance métabolique de l’erlotinib.
Trois voies métaboliques principales ont été identifiées : 1) O-déméthylation d’une ou des deux chaînes latérales, suivie d’une oxydation en acides carboxyliques ; 2) oxydation du groupement acétylène suivie d’une hydrolyse en acide arylcarboxylique et 3) hydroxylation aromatique du groupement phénylacétylène. Des dosages in vitro et des études de modèles tumoraux in vivo ont montré que les principaux métabolites de l’erlotinib, OSI-420 et OSI-413, produits par O-déméthylation de l’une ou l’autre des chaînes latérales exerçaient une activité similaire à celle de l’erlotinib. Ils sont présents dans le plasma à des concentrations inférieures à 10% de celles de l’erlotinib et leurs paramètres pharmacocinétique sont similaires à ce dernier.
Élimination
L'erlotinib est principalement excrété sous forme de métabolites dans les fèces (>90%), l’élimination rénale ne représentant qu’une faible proportion (environ 9%) d’une dose administrée par voie orale. Moins de 2% de la dose administrée oralement sont excrétés sous forme inchangée. Une analyse pharmacocinétique à l'échelon d'une population de 591 patients recevant l’erlotinib en monothérapie a montré une clairance moyenne apparente de 4,47 l/h et une demi-vie médiane de 36,2 heures. De ce fait, le délai d’obtention de l’état d’équilibre des concentrations plasmatiques devrait être voisin de 7-8 jours.
Pharmacocinétique dans des populations particulières :
En se basant sur les analyses de pharmacocinétique de population, aucune relation significative entre la clairance apparente prévue et l’âge, le poids, le sexe et l’origine ethnique des patients n'a été observée. Les facteurs liés au patient et corrélés aux paramètres pharmacocinétiques de l’erlotinib sont la bilirubinémie totale, la concentration en α-1GPA et être fumeur.
Des valeurs augmentées des concentrations plasmatiques de la bilirubine totale et de la concentration en α-1GPA ont été associées à une diminution de la clairance de l’erlotinib. La signification clinique de ces différences n’est pas claire. Toutefois, la clairance de l’erlotinib a été augmentée chez les fumeurs. Ceci a été confirmé par une étude pharmacocinétique chez des volontaires sains non-fumeurs ou fumeurs actifs traités par une dose orale unique de 150 mg d’erlotinib. La moyenne géométrique de la Cmax était de 1056 ng/mL chez les non-fumeurs et 689 ng/mL chez les fumeurs avec un rapport moyen de fumeurs à non-fumeurs de 65,2 % (IC95% : 44,3 à 95,9, p= 0,031). La moyenne géométrique de l’ASC0-inf était de 18726 ngh/mL chez les non-fumeurs et 6718 ngh/mL chez les fumeurs avec un rapport moyen de 35,9 % (IC95% : 23,7 à 54,3, p<0,0001). La moyenne géométrique de la C24h était de 288 ng/mL chez les non-fumeurs et 34,8 ng/mL chez les fumeurs avec un rapport moyen de 12,1 % (IC95% : 4,82 à 30,2, p= 0,0001).
Dans l’étude pivotale de phase III dans le CBNPC, les fumeurs actifs ont atteint l’état d’équilibre de l’erlotinib à une concentration plasmatique de 0,65 μg/mL (n=16) ce qui correspond à une concentration environ 2 fois inférieure à celle d’anciens fumeurs ou ceux qui n’ont jamais fumés (1,28 μg/mL, n=108). Cet effet était accompagné par une augmentation de 24% de la clairance plasmatique apparente de l’erlotinib. Dans une étude de phase I d’escalade de dose dans le CBNPC chez les patients fumeurs actifs, les analyses pharmacocinétiques à l’état d’équilibre ont montrés une augmentation dose dépendante de l’aire sous la courbe de l’erlotinib lorsque la posologie d’erlotinib était augmentée de 150 mg à la dose maximale tolérée de 300 mg. Dans cette étude, l’état d’équilibre des concentrations plasmatiques à une posologie de 300 mg chez les fumeurs actifs était de 1,22 μg/mL (n=17) (voir les rubriques 4.2, 4.4, 4.5 et 5.1).
Du fait des résultats des études de pharmacocinétique lors du traitement par l’erlotinib, les patients fumeurs devront être encouragés à arrêter, compte tenu de la réduction possible des concentrations plasmatiques d’erlotinib. Sur la base de l’étude de pharmacocinétique de population, il apparaît que la présence d’un opioïde augmente l’exposition d’environ 11%. Une seconde analyse de pharmacocinétique de population a été menée et a intégré des données sur l’erlotinib obtenues chez 204 patients atteints d’un cancer du pancréas ayant reçu l'erlotinib en association à la gemcitabine. Cette analyse a démontré que les covariables influençant la clairance de l'erlotinib chez les patients inclus dans l’étude menée dans le cancer du pancréas étaient très similaires à celles observées lors de l’analyse pharmacocinétique précédente en monothérapie. Aucun nouvel effet de covariance n'a été identifié. La co-administration avec la gemcitabine ne modifie pas la clairance plasmatique de l’erlotinib.
Population pédiatrique
Aucune étude n’a été spécifiquement menée en pédiatrie.
Population âgée
Aucune étude n’a été spécifiquement menée chez les personnes âgées.
Insuffisance hépatique
L’erlotinib est principalement éliminé par le foie. Chez les patients ayant des tumeurs solides et une insuffisance hépatique modérée (score de Child-Pugh 7-9), la moyenne géométrique de l’ASC0-t et la Cmax de l’erlotinib étaient respectivement de 27000 ngh/mL et 805 ng/mL comparées à 29300 ngh/mL et 1090 ng/mL chez les patients ayant une fonction hépatique adéquate y compris ceux ayant un cancer primitif du foie ou des métastases hépatiques. Bien que la Cmax soit statistiquement inférieure chez les patients avec une insuffisance hépatique modérée, cette différence n’est pas considérée comme cliniquement pertinente. Aucune donnée n’est disponible quant à l’influence de troubles fonctionnels hépatiques sévères sur les paramètres pharmacocinétiques de l’erlotinib. Sur la base des analyses de pharmacocinétique de population, l’augmentation des concentrations sériques en bilirubine totale était associée à une diminution de la clairance de l’erlotinib.
Insuffisance rénale
L’erlotinib et ses métabolites ne sont pas excrétés de façon significative par voie rénale. Moins de 9% d’une dose unique sont éliminés dans les urines Sur la base des analyses de pharmacocinétique de population, aucune relation cliniquement significative n’a été observée entre la clairance de l’erlotinib et la clairance de la créatinine. Mais, il n’y a pas de données disponibles chez les patients ayant une clairance de la créatinine <15 ml/min.
5.3. Données de sécurité préclinique
Les effets d’une administration chronique observés chez au moins une espèce animale ou lors d’une étude chez l’animal ont été notamment cornéens (atrophie, ulcération), cutanés (dégénérescence folliculaires et inflammation, rougeur et alopécie), ovariens (atrophie), hépatiques (nécrose du foie), rénaux (nécrose papillaire et dilatation tubulaire) et du tractus gastro-intestinal (retard de la vidange gastrique et diarrhée). Il a été observé une diminution des paramètres érythrocytaires et une augmentation des leucocytes, principalement des neutrophiles. Des élévations des concentrations en ALAT et ASAT et de la bilirubinémie ont été observées et reliées au traitement. Ces résultats ont été observés à des expositions bien inférieures à celles cliniquement significatives.
Du fait de son mode d’action, l'erlotinib a un potentiel tératogène. Des données issues d’études de la toxicité sur la reproduction menées chez le rat et le lapin, à des doses voisines de la dose maximale tolérée (DMT) et/ou toxiques pour les mères, ont reporté une toxicité de reproduction (embryotoxicité chez les rats, résorption embryonnaire et foetotoxicité chez les lapins) et une toxicité de développement (diminution de la croissance et de la survie chez les jeunes rats), mais n’ont révélé aucun signe de tératogénicité ou d’altération de la fertilité. Ces résultats ont été observés à des expositions cliniquement significatives.
Les études de génotoxicité conventionnelles menées avec l’erlotinib se sont révélées négatives. Des études de carcinogénicité de 2 ans réalisées chez des rats et des souris à des niveaux d’exposition supérieurs au niveau d’exposition thérapeutique humain (respectivement, jusqu’à 2 fois et 10 fois supérieurs en se basant sur la Cmax et/ou l’ASC) ont été négatives.
Une réaction cutanée phototoxique modérée a été observée chez les rats après irradiation par les UV.
Lactose monohydraté, cellulose microcristalline (E460), carboxyméthylamidon sodique Type A, stéarate de magnésium (E470 b)
Pelliculage du comprimé :
Poly(alcool vinylique) (E1203), dioxyde de titane (E171), macrogol 3350 (E1521), talc (E553b), copolymère d’acide méthacrylique et d’acrylate d’éthyle (1:1) Type A, bicarbonate de sodium.
4 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Plaquettes (Aluminium-OPA/Aluminium/PVC) de 30 comprimés, emballées dans des boîtes en carton.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières pour l’élimination.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
15 boulevard Charles de Gaulle
92700 Colombes
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 115 3 6 : 30 comprimés sous plaquettes (Aluminium -OPA/Aluminium/PVC)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en oncologie ou en hématologie ou aux médecins compétents en cancérologie.
ANSM - Mis à jour le : 16/08/2023
ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé
Erlotinib
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé ?
3. Comment prendre ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : agent antinéoplasique inhibiteur de protéine kinase - code ATC: L01EB02.
ERLOTINIB BIOGARAN contient une substance active appelée erlotinib. ERLOTINIB BIOGARAN est un médicament destiné à traiter le cancer en bloquant l’activité d’une protéine appelée récepteur du facteur de croissance épidermique (EGFR). On sait que cette protéine intervient dans la croissance et la dissémination des cellules cancéreuses.
ERLOTINIB BIOGARAN est indiqué chez l’adulte. Ce médicament peut vous être prescrit si vous souffrez d’un cancer du poumon non à petites cellules à un stade avancé. Il peut vous être prescrit en traitement initial ou en traitement si votre maladie reste inchangée après une chimiothérapie initiale sous réserve que les cellules de votre cancer aient des mutations de l’EGFR spécifiques. Il peut également vous être prescrit si la progression n’a pas été arrêtée par une chimiothérapie précédente. Ce médicament peut vous être également prescrit en association à un autre médicament appelé gemcitabine si vous souffrez d’un cancer du pancréas à un stade métastatique.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé ?
Ne prenez jamais ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé :
· si vous êtes allergique à l’erlotinib ou à l’un des autres composants contenus dans ce médicament (mentionnés à la rubrique 6).
Avertissements et précautions
· si vous prenez d’autres médicaments qui peuvent augmenter ou diminuer la quantité d’erlotinib dans votre sang ou influencer son effet (par exemple les antifongiques comme le kétoconazole, les inhibiteurs de protéases, l’érythromycine, la clarithromycine, la phénytoïne, la carbamazépine, les barbituriques, la rifampicine, la ciprofloxacine, l’oméprazole, la ranitidine, le millepertuis ou les inhibiteurs du protéasome), parlez-en à votre médecin. Dans certains cas, ces médicaments peuvent diminuer l’efficacité ou augmenter les effets indésirables de ERLOTINIB BIOGARAN et votre médecin pourra décider qu’un ajustement de votre traitement est nécessaire. Votre médecin devrait éviter de vous traiter avec ces médicaments pendant votre traitement par ERLOTINIB BIOGARAN.
· si vous prenez des anticoagulants (médicaments qui participent à la prévention des thromboses ou de la coagulation sanguine, comme par exemple la warfarine), ERLOTINIB BIOGARAN peut augmenter votre tendance à saigner. Parlez-en à votre médecin, il devra vous suivre régulièrement au moyen d’analyses sanguines.
· si vous prenez des statines (médicaments qui diminuent le cholestérol dans le sang), ERLOTINIB BIOGARAN peut augmenter le risque de troubles musculaires induits par les statines, qui dans de rares cas peuvent mener à une dégradation sévère des muscles (rhabdomyolyse) conduisant à une lésion du rein, parlez-en à votre médecin.
· si vous utilisez des lentilles de contact et/ou si avez eu par le passé des problèmes au niveau des yeux comme une sécheresse sévère des yeux, une inflammation de la partie avant de l’œil (cornée) ou des ulcères au niveau de la partie avant de l’œil, parlez-en à votre médecin.
Reportez-vous également ci-dessous à « Autres médicaments et ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé ».
Veuillez indiquer à votre médecin :
· si vous ressentez des difficultés respiratoires soudaines associées à une toux ou une fièvre, car votre médecin peut alors décider de vous traiter par d’autres médicaments et d’interrompre votre traitement par ERLOTINIB BIOGARAN ;
· si vous avez une diarrhée, car votre médecin peut alors décider de vous prescrire un antidiarrhéique (par exemple le lopéramide) ;
· immédiatement, si vous avez, de façon sévère et persistante, une diarrhée, des nausées, une perte d’appétit ou des vomissements, car votre médecin peut alors décider d’interrompre votre traitement par ERLOTINIB BIOGARAN et pourrait avoir besoin de vous hospitaliser ;
· si vous avez des douleurs abdominales sévères, des cloques importantes ou une desquamation sévère de la peau. Votre médecin peut décider d’interrompre ou d’arrêter définitivement votre traitement ;
· si vous présentez une rougeur ou une douleur de l’œil, aiguë ou qui s’aggrave, une augmentation de larmoiement, une vision floue et/ou une sensibilité à la lumière, parlez en immédiatement à votre médecin ou infirmier/ère car vous pourriez avoir besoin d’un traitement d’urgence (reportez-vous ci-dessous à « Quels sont les effets indésirables éventuels »).
· si vous prenez une statine ou si vous avez des douleurs musculaires inexpliquées, un endolorissement, une faiblesse musculaire ou des crampes. Votre médecin peut décider d’interrompre ou d’arrêter définitivement votre traitement.
Reportez-vous également à la rubrique 4 « Quels sont les effets indésirables éventuels ».
Maladie du foie ou des reins
On ne sait pas si l’effet de ERLOTINIB BIOGARAN est différent en cas d’anomalie du fonctionnement de votre foie ou de vos reins. Ce traitement n’est pas recommandé si vous souffrez de maladie sévère du foie ou du rein.
Trouble de la glucuroconjugaison comme la maladie de Gilbert
Votre médecin doit vous traiter avec précaution si vous présentez des troubles de la glucuroconjugaison comme la maladie de Gilbert.
Fumeurs
Il vous est conseillé d’arrêter de fumer si vous êtes traité par ERLOTINIB BIOGARAN car fumer peut entraîner une diminution de la quantité de ce médicament dans le sang.
Enfants et adolescents
ERLOTINIB BIOGARAN n’a pas été étudié chez les patients âgés de moins de 18 ans. Ce traitement est déconseillé pour les enfants et les adolescents.
Autres médicaments et ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé
Si vous prenez, si vous avez pris récemment ou si vous pourriez prendre un autre médicament, parlez-en à votre médecin ou à votre pharmacien.
ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé avec des aliments et boissons
Ne prenez pas ERLOTINIB BIOGARAN en même temps que des aliments. Veuillez-vous reporter également à la rubrique 3 « Comment prendre ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé ».
Evitez une grossesse pendant le traitement par ERLOTINIB BIOGARAN. Si vous êtes en âge de procréer, utilisez une méthode de contraception adéquate pendant le traitement et pendant au moins 2 semaines après la prise du dernier comprimé. Si vous débutez une grossesse pendant le traitement par ERLOTINIB BIOGARAN informez immédiatement votre médecin, qui décidera si le traitement doit être poursuivi.
N’allaitez pas pendant le traitement par ERLOTINIB BIOGARAN et pendant au moins 2 semaines après la prise du dernier comprimé.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Les effets de ERLOTINIB BIOGARAN sur l’aptitude à conduire des véhicules et à utiliser des machines n’ont pas été étudiés. Toutefois, il est très peu probable que votre traitement puisse les affecter.
ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé contient un sucre appelé lactose monohydraté et du sodium.
Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé ?
Les comprimés doivent être pris au moins une heure avant ou deux heures après l’ingestion de nourriture.
Si vous souffrez d’un cancer du poumon non à petites cellules, la dose habituelle de ERLOTINIB BIOGARAN est de 150 mg chaque jour.
Si vous souffrez d’un cancer du pancréas métastatique, la dose habituelle de ERLOTINIB BIOGARAN est de 100 mg par jour. ERLOTINIB BIOGARAN est associé à un autre médicament, la gemcitabine.
Votre médecin peut ajuster votre posologie par palier de 50 mg. Pour les adaptations de posologie, ERLOTINIB BIOGARAN est disponible en dosages de 25 mg, 100 mg ou 150 mg.
Si vous avez pris plus de ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé que vous n’auriez dû
Contactez immédiatement votre médecin ou votre pharmacien.
Vous pouvez rencontrer des effets indésirables plus importants et votre médecin peut interrompre votre traitement.
Si vous oubliez de prendre ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé
Si vous avez oublié de prendre une ou plusieurs doses de ERLOTINIB BIOGARAN, contactez votre médecin ou votre pharmacien dès que possible. Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé
Il est important de prendre ERLOTINIB BIOGARAN tous les jours tant que votre médecin vous le prescrit.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Contactez votre médecin dès que possible si vous souffrez d’un des effets indésirables ci-dessous. Dans certains cas, votre médecin pourra décider de réduire votre dose de ERLOTINIB BIOGARAN ou d’interrompre le traitement :
· Diarrhée et vomissement (très fréquent : qui peut toucher plus d’une personne sur 10). Une diarrhée persistante et sévère peut conduire à une baisse du potassium dans le sang et à une altération du fonctionnement du rein, en particulier si vous recevez d’autres traitements de chimiothérapie en même temps. Si vous avez une diarrhée plus sévère et persistante, contactez votre médecin immédiatement car une hospitalisation pourrait être nécessaire.
· Irritation des yeux due à une kératoconjonctivite (très fréquent : qui peut toucher plus d’une personne sur 10), une conjonctivite et une kératite (fréquent : qui peut toucher au maximum une personne sur 10).
· Une forme d’irritation des poumons appelée affection pulmonaire interstitielle (peu fréquent chez les patients européens : qui peut toucher au maximum une personne sur 100 ; fréquent chez les patients japonais : qui peut toucher au maximum une personne sur 10). Cette maladie peut être également liée à la progression naturelle de votre maladie et peut être fatale dans certains cas. Si vous ressentez des symptômes tels que des difficultés subites à respirer avec une toux ou une fièvre, contactez immédiatement votre médecin car vous pourriez souffrir de cette affection. Votre médecin peut décider d’arrêter définitivement votre traitement par ERLOTINIB BIOGARAN.
· Des perforations gastro-intestinales ont été observées (peu fréquent : qui peut toucher au maximum une personne sur 100). Indiquez à votre médecin si vous avez des douleurs abdominales sévères. Indiquez également à votre médecin si vous avez eu par le passé des ulcères à l’estomac ou une maladie diverticulaire, car ce risque peut être augmenté.
· Dans de rares cas, une inflammation du foie (hépatite) a été observée (peut toucher au maximum une personne sur 1 000). Les symptômes peuvent inclure une sensation générale de malaise, avec ou sans possible jaunisse (jaunissement de la peau et des yeux), des urines foncées, des nausées, des vomissements et des douleurs abdominales. Dans de rares cas, une insuffisance hépatique a été observée. Elle peut être potentiellement fatale. Si vos analyses de sang montrent une modification importante du fonctionnement de votre foie, votre médecin pourrait devoir interrompre votre traitement.
Effets indésirables très fréquents (qui peuvent toucher plus d’une personne sur 10) :
· Eruption cutanée qui peut survenir ou s’aggraver au niveau des régions exposées au soleil.
Si vous vous exposez au soleil, des vêtements protecteurs, et/ou une protection solaire (par exemple à base de filtres minéraux) pourraient être recommandés.
· Infection
· Perte d’appétit, perte de poids
· Dépression
· Maux de tête, altération des sensations tactiles ou engourdissement des membres
· Difficulté à respirer, toux
· Nausée
· Irritation de la bouche
· Maux d’estomac, indigestion et flatulences
· Anomalies des analyses sanguines mesurant le fonctionnement du foie
· Démangeaisons
· Fatigue, fièvre, frissons.
Effets indésirables fréquents (qui peuvent toucher au maximum une personne sur 10)
· Peau sèche
· Perte de cheveux
· Saignements de nez
· Saignements de l’estomac ou des intestins
· Réactions inflammatoires autour de l’ongle du doigt
· Infection des follicules pileux
· Acné
· Peau craquelée (fissures de la peau)
· Altération de la fonction rénale (lorsque le médicament est pris en dehors des indications approuvées en association avec une chimiothérapie).
Effets indésirables peu fréquents (qui peuvent toucher au maximum une personne sur 100)
· Inflammation des reins (néphrite)
· Excès de protéines dans les urines (protéinurie)
· Des modifications des cils
· Des augmentations de la pilosité au niveau du corps et du visage avec une distribution androgénique
· Excès de pigmentation de la peau
· Des modifications des sourcils
· Des ongles cassants et une perte des ongles.
Effets indésirables rares (qui peuvent toucher au maximum une personne sur 1000) :
· Rougeur ou douleur au niveau de la paume des mains ou de la plante des pieds (Syndrome d’érythrodysesthésie palmo-plantaire).
Effets indésirables très rares (qui peuvent toucher au maximum une personne sur 10000)
· Des cas d’ulcération ou de perforation de la cornée
· Des vésicules ou des desquamations importantes de la peau (suggérant un syndrome de Stevens-Johnson)
· Inflammation de la partie colorée de l’œil.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou, votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé
· La substance active est :
Erlotinib........................................................................................................................ 150 mg
sous forme de chlorhydrate d'erlotinib
Pour un comprimé pelliculé.
· Les autres composants sont :
Noyau du comprimé :
Lactose monohydraté, cellulose microcristalline (E460), carboxyméthylamidon sodique Type A, stéarate de magnésium (E470 b)
Pelliculage du comprimé :
Poly(alcool vinylique) (E1203), dioxyde de titane (E171), macrogol 3350 (E1521), talc (E553b), copolymère d’acide méthacrylique et d’acrylate d’éthyle (1:1), Type A, bicarbonate de sodium
Qu’est-ce que ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé et contenu de l’emballage extérieur
ERLOTINIB BIOGARAN 150 mg, comprimé pelliculé se présente sous forme de comprimé blanc à jaunâtre, rond, biconvexe, pelliculé, avec gravure « 150 » sur une face, de diamètre 10,5 mm ± 5 %.
Les comprimés sont présentés sous plaquettes (Aluminium-OPA/Aluminium/PVC) de 30 comprimés, emballées dans des boîtes en carton.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
BIOGARAN
15 BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
Exploitant de l’autorisation de mise sur le marché
15 BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
REMEDICA LTD
AHARNON STREET
LIMASSOL INDUSTRIAL ESTATE
3056 LIMASSOL
CHYPRE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).