Dernière mise à jour le 28/04/2026
METOJECT 17,5 mg/0,35 ml, solution injectable en stylo prérempli
Indications thérapeutiques
Classe pharmacothérapeutique – code ATC : autres immunosuppresseurs - L04AX03.
METOJECT est indiqué dans le traitement :
· de la polyarthrite rhumatoïde sévère et active chez l’adulte ;
· des formes polyarticulaires de l’arthrite juvénile idiopathique active sévère, en cas de réponse inadéquate aux anti‑inflammatoires non stéroïdiens (AINS) ;
· des formes modérées à sévères du psoriasis chez les patients adultes et des formes sévères du rhumatisme psoriasique chez l’adulte ;
· des formes légères à modérées de la maladie de Crohn chez l’adulte lorsqu’un traitement adéquat par d’autres médicaments n’est pas possible.
La polyarthrite rhumatoïde (PR) est une maladie chronique du collagène caractérisée par une inflammation des membranes synoviales (membranes des articulations). Ces membranes sécrètent un liquide qui agit comme un lubrifiant pour de nombreuses articulations. L’inflammation provoque un épaississement de la membrane et un gonflement de l’articulation.
L’arthrite juvénile est une maladie qui touche les enfants et adolescents de moins de 16 ans. On parle de formes polyarticulaires lorsque 5 articulations ou plus sont touchées au cours des six premiers mois de la maladie.
Le psoriasis est une affection cutanée chronique courante caractérisée par des plaques rouges recouvertes de squames adhérentes nacrées, épaisses et sèches.
Le rhumatisme psoriasique est une forme d’arthrite accompagnée de lésions psoriasiques de la peau et des ongles, notamment au niveau des articulations des doigts et des orteils.
METOJECT modifie et ralentit la progression de la maladie.
La maladie de Crohn est un type de maladie inflammatoire des intestins qui peut affecter n’importe quelle région du tube gastro-intestinal et provoque des symptômes tels que des douleurs abdominales, de la diarrhée, des vomissements ou une perte de poids.
Présentations
> 1 stylo prérempli de 0,35 ml avec seringue(s) en verre avec aiguille(s)
Code CIP : 34009 300 203 6 4
Déclaration de commercialisation : 26/10/2016
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 16,54 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 17,56 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 05/02/2020 | Extension d'indication | Le service médical rendu par METOJECT (méthotrexate) est important dans le traitement des formes modérées de psoriasis chez les adultes candidats à un traitement systémique et aux posologies de l’AMM. |
| Important | Avis du 12/12/2018 | Renouvellement d'inscription (CT) | Le service médical rendu par METOJECT reste important dans les indications et aux posologies de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 05/02/2020 | Extension d'indication | Compte tenu : • de la démonstration de la supériorité du méthotrexate injectable par rapport au placebo chez des patients adultes ayant un psoriasis modéré à sévère en termes de pourcentage de répondeurs PASI 75, candidats à un traitement systémique, • de l’absence de comparaison à d’autres traitements systémiques de 1ère intention, la Commission de la transparence considère que METOJECT (méthotrexate) n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres traitement systémiques de 1ère intention dans la prise en charge des formes modérées du psoriasis chez les adultes candidats à un traitement systémique. |
| V (Inexistant) | Avis du 16/03/2016 | Inscription (CT) | Ces spécialités sont des compléments de gamme qui n’apportent pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres présentations de METOJECT déjà inscrites. |
ANSM - Mis à jour le : 15/10/2024
METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Méthotrexate . .. 17,5 mg
Pour un stylo prérempli avec 0,35 ml de solution.
1 ml de solution injectable contient 50 mg de méthotrexate.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable en stylo prérempli.
Solution limpide, de couleur brun-jaune.
4.1. Indications thérapeutiques
METOJECT est indiqué dans le traitement :
· de la polyarthrite rhumatoïde sévère et active chez l’adulte ;
· des formes polyarticulaires de l’arthrite juvénile idiopathique active sévère, en cas de réponse inadéquate aux anti‑inflammatoires non stéroïdiens (AINS) ;
· des formes modérées à sévères du psoriasis chez les patients adultes candidats à un traitement systémique et des formes sévères du rhumatisme psoriasique chez l’adulte ;
· des formes légères à modérées de la maladie de Crohn, seul ou en association avec des corticostéroïdes, chez les patients adultes réfractaires ou intolérants aux thiopurines.
4.2. Posologie et mode d'administration
Avertissement important concernant l’administration de METOJECT (méthotrexate)
Dans le traitement de la polyarthrite rhumatoïde, de l’arthrite juvénile idiopathique, du psoriasis, du rhumatisme psoriasique et de la maladie de Crohn, METOJECT (méthotrexate) doit être utilisé une fois par semaine seulement. Des erreurs de posologie lors de l’utilisation de METOJECT (méthotrexate) peuvent entraîner des effets indésirables graves, voire le décès du patient. Veuillez lire très attentivement cette rubrique du résumé des caractéristiques du produit.
Le méthotrexate ne doit être prescrit que par un médecin ayant de l’expertise dans l’utilisation du méthotrexate et une compréhension exhaustive des risques du traitement par méthotrexate. Les patients doivent être instruits et formés à la technique d’injection adaptée lorsqu’ils s’auto-administrent du méthotrexate. La première injection de METOJECT doit être réalisée sous surveillance médicale directe. METOJECT est injecté une fois par semaine.
Le patient doit être informé de façon claire du mode d’administration hebdomadaire de METOJECT. Il est recommandé de choisir un jour de la semaine approprié comme jour d’injection à respecter chaque semaine.
L’élimination du méthotrexate est diminuée chez les patients présentant un espace de distribution tiers (ascite, épanchement pleural). Chez ces patients, il convient de surveiller étroitement les signes de toxicité et il peut être nécessaire de diminuer la posologie, voire, dans certains cas, d’arrêter le traitement par méthotrexate (voir rubriques 5.2 et 4.4).
Posologie
Traitement de la polyarthrite rhumatoïde chez l’adulte
La dose initiale recommandée est de 7,5 mg de méthotrexate une fois par semaine, administrée par voie sous-cutanée. En fonction de l’activité individuelle de la maladie et de la tolérance du patient, la dose initiale pourra être augmentée progressivement de 2,5 mg par semaine.
Il ne faut pas dépasser une dose hebdomadaire de 25 mg. Les doses supérieures à 20 mg/semaine peuvent être associées à une augmentation significative de la toxicité, notamment de l’aplasie médullaire. La réponse au traitement est généralement observée après 4 à 8 semaines de traitement environ. Après obtention du résultat thérapeutique souhaité, la posologie doit être diminuée progressivement jusqu’à la dose d’entretien efficace la plus faible possible.
Population pédiatrique
Traitement des formes polyarticulaires de l’arthrite juvénile idiopathique chez l’enfant et l’adolescent de moins de 16 ans
La dose recommandée est de 10 à 15 mg/m² de surface corporelle (SC) une fois par semaine. Dans les cas réfractaires, la posologie hebdomadaire pourrait être augmentée jusqu’à 20 mg/m² de surface corporelle une fois par semaine. Toutefois, une surveillance plus fréquente serait alors recommandée.
Compte tenu des données limitées concernant l’administration intraveineuse chez l’enfant et l’adolescent, l’administration parentérale doit être limitée à la voie sous-cutanée.
Les patients atteints d’AJI doivent toujours être adressés à un rhumatologue spécialisé dans le traitement des patients pédiatriques.
Ce médicament ne doit pas être utilisé chez l’enfant de moins de 3 ans compte tenu de l’insuffisance de données concernant la sécurité et l’efficacité du produit dans cette population (voir rubrique 4.4).
Traitement du psoriasis en plaques et du rhumatisme psoriasique
Il est recommandé d’administrer une dose test de 5 à 10 mg par voie parentérale une semaine avant le début du traitement pour détecter des effets indésirables idiosyncrasiques. La dose initiale recommandée est de 7,5 mg de méthotrexate une fois par semaine, administrée par voie sous-cutanée. La posologie doit être augmentée progressivement mais en général, elle ne doit pas excéder 25 mg de méthotrexate par semaine. Les doses supérieures à 20 mg/semaine peuvent être associées à une augmentation significative de la toxicité, notamment de l’aplasie médullaire. La réponse au traitement est généralement observée dans les 2 à 6 semaines environ suivant le début du traitement. Après obtention du résultat thérapeutique souhaité, la posologie doit être diminuée progressivement jusqu’à la dose d’entretien efficace la plus faible possible.
Dose hebdomadaire maximale
La dose doit être augmentée selon les besoins, mais en général, elle ne doit pas excéder la dose hebdomadaire maximale recommandée de 25 mg. Dans certains cas exceptionnels, une dose plus élevée peut être cliniquement justifiée, mais elle ne doit pas excéder une dose hebdomadaire maximale de 30 mg de méthotrexate du fait de l’augmentation notable de la toxicité.
Traitement de la maladie de Crohn
Traitement d’induction :
25 mg/semaine par voie sous-cutanée.
La réponse au traitement peut être attendue après 8 à 12 semaines environ.
Traitement d’entretien :
15 mg/semaine par voie sous-cutanée.
L’expérience chez la population pédiatrique n’est pas suffisante pour que puisse être recommandée l’utilisation de METOJECT pour le traitement de la maladie de Crohn chez cette population.
Patients insuffisants rénaux
METOJECT doit être utilisé avec prudence chez les patients présentant une insuffisance rénale. La posologie doit être adaptée comme suit :
Clairance de la créatinine (ml/min) Dose
≥ 60 100 %
30 – 59 50 %
< 30 Ne pas utiliser METOJECT
Voir la rubrique 4.3.
Patients insuffisants hépatiques
Le méthotrexate doit être administré avec une grande prudence, voire évité, chez les patients présentant ou ayant des antécédents d’hépatopathie sévère, en particulier d’origine éthylique. Le méthotrexate est contre-indiqué chez les patients ayant un taux de bilirubine > 5 mg/dl (85,5 µmol/l).
Pour la liste complète des contre-indications, voir rubrique 4.3.
Patients âgés
Une réduction de la posologie doit être envisagée chez les patients âgés en raison de la diminution des fonctions hépatique et rénale et des réserves d’acide folique plus faibles chez ces patients.
Patients présentant un espace de distribution tiers (épanchement pleural, ascite)
La demi-vie du méthotrexate pouvant être prolongée jusqu’à 4 fois la valeur normale chez les patients qui présentent un espace de distribution tiers, il peut être nécessaire de diminuer la posologie voire, dans certains cas, d’arrêter le traitement (voir rubriques 5.2 et 4.4).
Mode d'administration
Ce médicament est à usage unique exclusivement.
METOJECT, solution injectable en stylo prérempli, peut uniquement être administré par voie sous-cutanée.
La durée globale du traitement doit être déterminée par le médecin.
Des instructions concernant l’utilisation de METOJECT, solution injectable en stylo prérempli sont disponibles ci‑dessous.
Notons que l’intégralité du contenu doit être utilisée lors de chaque administration.
Instructions pour l’utilisation sous‑cutanée de METOJECT avec capuchon jaune et bouton poussoir jaune (auto‑injecteur en trois étapes)
Les zones les plus appropriées pour l’injection du produit sont :
· le haut des cuisses
· le ventre, à distance du nombril.
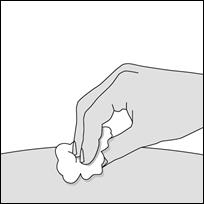
1. Nettoyez la zone choisie pour l’injection.
2. Ôtez le capuchon du stylo en tirant d’un coup sec dans l’axe du stylo.
3. Formez un pli cutané entre le pouce et l’index sur la zone choisie pour l’injection.
4. Maintenez le pli pendant toute la durée de l’injection et ne relâchez la peau qu’une fois l’injection terminée et le stylo retiré.
5. Appuyez le stylo METOJECT sur la peau et selon un angle de 90 degrés afin de déverrouiller le bouton poussoir. Appuyez ensuite sur le bouton poussoir (un déclic indique le début de l’injection).
6. Ne pas retirez METOJECT avant d’avoir terminé l’injection, afin de garantir que la totalité du produit a bien été injectée. La durée de l’injection peut atteindre les 5 secondes.
7. Retirez METOJECT, toujours en maintenant le stylo selon un angle de 90 degrés par rapport à la surface cutanée.
8. L’embout de sécurité se remet en place autour de l’aiguille et se verrouille automatiquement.
Instructions pour l’utilisation sous‑cutanée de METOJECT avec capuchon de protection translucide et protège‑aiguille bleu (auto‑injecteur en deux étapes)
Les zones les plus appropriées pour l’injection sont :
· le haut des cuisses,
· le bas du ventre, en évitant la zone de 5 cm autour du nombril.
1. Nettoyez le site d’injection.
2. Retirez le capuchon translucide en tirant d’un coup sec dans l’axe du stylo.
3. Placez le protège‑aiguille bleu décapuchonné sur la peau à la verticale (90 degrés). Il n’est pas nécessaire de former un pli cutané.
4. Commencez l’injection en pressant le stylo METOJECT à fond contre la peau (un premier déclic indique le début de l’injection).
5. Maintenez METOJECT contre la peau jusqu’à la fin de l’injection, qui est terminée lorsque :
ü vous entendez un deuxième déclic, peu après le premier ;
ü ou : la tige bleue du piston a cessé de se déplacer et remplit la fenêtre d’inspection ;
ü ou : 5 secondes se sont écoulées.
6. Retirez METOJECT de la peau, en le tenant toujours à la verticale (90 degrés).
7. Le protège‑aiguille bleu se met automatiquement en place sur l’aiguille et se verrouille alors.
Remarque :
En cas de passage de la voie orale à une administration parentérale, il peut être nécessaire de diminuer la posologie du fait de la biodisponibilité variable du méthotrexate après administration orale.
Une supplémentation en acide folique peut être envisagée conformément aux recommandations thérapeutiques en vigueur.
METOJECT est contre-indiqué dans les cas suivants :
· hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1 ;
· troubles sévères de la fonction hépatique (voir rubrique 4.2) ;
· alcoolisme ;
· troubles sévères de la fonction rénale (clairance de la créatinine inférieure à 30 ml/min, voir rubrique 4.2 et rubrique 4.4) ;
· dyscrasies sanguines préexistantes, telles qu’une hypoplasie médullaire, une leucopénie, une thrombopénie ou une anémie sévère ;
· infections graves, aiguës ou chroniques telles que tuberculose, infection par le VIH ou autres syndromes d’immunodéficience ;
· ulcères de la cavité buccale ou maladie ulcéreuse gastro-intestinale évolutive avérée ;
· grossesse et allaitement (voir rubrique 4.6) ;
· administration concomitante de vaccins vivants.
4.4. Mises en garde spéciales et précautions d'emploi
Les patients sous traitement doivent faire l’objet d’une surveillance appropriée visant à détecter et évaluer le plus tôt possible d’éventuels signes de toxicité ou effets indésirables. Par conséquent, un traitement par méthotrexate doit uniquement être instauré et supervisé par un médecin connaissant bien les traitements par antimétabolites et ayant l’expérience de leur utilisation. Étant donné le risque de réactions toxiques sévères voire fatales, le patient doit être pleinement informé par le médecin des risques encourus et des mesures de sécurité recommandées.
Examens et mesures de sécurité recommandés
Avant l’instauration ou la réintroduction du traitement par le méthotrexate après une période de repos
Numération de la formule sanguine et plaquettes (NFS), enzymes hépatiques, bilirubine, albuminémie, radiographie thoracique et tests de la fonction rénale. Si cela est cliniquement justifié, des examens doivent être pratiqués pour exclure la présence d’une tuberculose ou d’une hépatite.
Pendant le traitement
Les examens suivants doivent être réalisés une fois par semaine au cours des deux premières semaines puis toutes les deux semaines pendant le mois suivant ; ensuite, en fonction de la numération leucocytaire et de la stabilité du patient, au moins une fois par mois durant les six mois qui suivent et au moins tous les trois mois par la suite.
Une augmentation de la fréquence de suivi doit également être envisagée lors d’une augmentation de dose. Les patients âgés, en particulier, doivent être examinés fréquemment pour détecter les signes précoces de toxicité.
· Examen de la bouche et de la gorge visant à détecter des modifications des muqueuses.
· Numération de la formule sanguine comprenant la numération différentielle et de plaquettes. L’inhibition de l’hématopoïèse causée par le méthotrexate peut survenir subitement et à des doses habituellement sûres. Toute chute sévère du taux de leucocytes ou de plaquettes impose l’arrêt immédiat du médicament et la mise en place d’un traitement symptomatique approprié. Les patients doivent être incités à signaler tous les signes ou symptômes évocateurs d’une infection. La numération de la formule sanguine et les plaquettes doivent être surveillées étroitement chez les patients recevant de façon concomitante des médicaments hématotoxiques (léflunomide par exemple).
· Tests de la fonction hépatique : le traitement ne doit pas être instauré ou doit être interrompu en cas d’anomalies persistantes ou significatives des résultats des tests de la fonction hépatique, d’autres investigations non invasives d’une fibrose hépatique ou des biopsies hépatiques.
Des élévations transitoires des transaminases jusqu’à deux ou trois fois la limite supérieure de la normale ont été observées à une fréquence de 13 % à 20 %. Une élévation persistante des enzymes hépatiques et/ou une diminution de l’albuminémie peuvent indiquer une hépatotoxicité sévère. En cas d’augmentation persistante des enzymes hépatiques, il convient d’envisager une réduction de la dose ou l’arrêt du traitement.
Les modifications histologiques, la fibrose, et, plus rarement, la cirrhose, peuvent ne pas être précédées d’anomalies des résultats des tests de la fonction hépatique. Il existe des cas de cirrhose dans lesquels les valeurs des transaminases sont normales. Par conséquent, des méthodes diagnostiques non invasives pour la surveillance de l’état hépatique, en plus des tests de la fonction hépatique, doivent être envisagées. La biopsie hépatique doit être envisagée au cas par cas en prenant en compte les comorbidités et les antécédents médicaux du patient et les risques liés à la biopsie. Les facteurs de risque d’hépatotoxicité comprennent des antécédents de consommation excessive d’alcool, une élévation persistante des enzymes hépatiques, des antécédents d’hépatopathie, des antécédents familiaux de maladie hépatique héréditaire, le diabète, l’obésité, une exposition antérieure à des médicaments ou substances chimiques hépatotoxiques et un traitement de longue durée par le méthotrexate.
D’autres médicaments hépatotoxiques ne doivent être co-administrés qu’en cas de nécessité absolue pendant le traitement par le méthotrexate. La consommation d’alcool doit être évitée (voir rubriques 4.3 et 4.5). Une surveillance plus étroite des enzymes hépatiques doit être effectuée chez les patients recevant de façon concomitante d’autres médicaments hépatotoxiques.
Des précautions particulières s’imposent chez les patients atteints de diabète insulinodépendant, car des cas isolés de développement d’une cirrhose sans augmentation des enzymes hépatiques pendant le traitement par le méthotrexate ont été rapportés.
· La fonction rénale doit être surveillée par des tests de la fonction rénale et des analyses d’urine (voir rubriques 4.2 et 4.3).
Le méthotrexate étant éliminé essentiellement par voie rénale, une élévation des concentrations sériques pouvant entraîner des effets indésirables sévères peut être attendue en cas de troubles de la fonction rénale.
La surveillance doit être plus fréquente chez les patients dont la fonction rénale peut être altérée (par exemple chez les sujets âgés). Cette précaution s’impose en particulier en cas d’administration concomitante de médicaments qui affectent l’élimination du méthotrexate, qui provoquent une atteinte rénale (anti-inflammatoires non stéroïdiens par exemple) ou qui sont susceptibles de diminuer l’hématopoïèse. La déshydratation peut également majorer la toxicité du méthotrexate.
· Évaluation de l’appareil respiratoire : il convient de surveiller l’apparition des symptômes d’une insuffisance respiratoire et si nécessaire, de pratiquer des tests de la fonction pulmonaire. Une affection pulmonaire doit être diagnostiquée rapidement et impose l’arrêt du méthotrexate. L’apparition de symptômes pulmonaires (en particulier une toux sèche non productive) ou d’une pneumopathie non spécifique pendant le traitement par le méthotrexate peut être le signe d’une lésion potentiellement dangereuse et impose l’interruption du traitement et des investigations approfondies. Il existe un risque de pneumonie interstitielle aiguë ou chronique, souvent associée à une éosinophilie sanguine, et des cas fatals ont été rapportés. Bien que le tableau clinique soit variable, les symptômes typiques de la pneumopathie induite par le méthotrexate sont une fièvre, une toux, une dyspnée, une hypoxémie et la présence d’un infiltrat à la radiographie thoracique ; la présence d’une infection doit être exclue. Les maladies pulmonaires induites par le méthotrexate ne sont pas toujours totalement réversibles. Cette lésion peut survenir à tous les dosages.
En outre, des cas d’hémorragie alvéolaire pulmonaire ont été rapportés lorsque le méthotrexate est utilisé pour des indications rhumatologiques et apparentées. Cette affection peut également être associée à une vasculite et à d’autres comorbidités. Des examens doivent être rapidement envisagés en cas de suspicion d’hémorragie alvéolaire pulmonaire afin de confirmer le diagnostic.
· En raison de ses effets sur le système immunitaire, le méthotrexate peut diminuer la réponse aux vaccinations et modifier les résultats des tests immunologiques. Les patients présentant des infections chroniques inactives (par exemple zona, tuberculose, hépatite B ou C) doivent faire l’objet d’une attention particulière en raison d’une activation possible de l’infection. L’administration de vaccins vivants est contre-indiquée pendant le traitement par le méthotrexate.
Des lymphomes malins peuvent survenir chez les patients recevant le méthotrexate à faible dose ; dans ce cas, le traitement doit être arrêté. L’absence de signes de régression spontanée du lymphome impose l’instauration d’une chimiothérapie cytotoxique.
De rares cas de pancytopénie mégaloblastique aiguë ont été rapportés lors de l’administration concomitante d’antagonistes de l’acide folique tels que le triméthoprime-sulfaméthoxazole.
Photosensibilité
Une photosensibilité se manifestant par une réaction d’exanthème solaire exagérée a été observée chez certaines personnes prenant du méthotrexate (voir rubrique 4.8). Sauf indication médicale, l’exposition au soleil intense ou aux rayons UV doit être évitée. Les patients doivent utiliser une protection solaire adéquate pour se protéger du soleil intense.
Les dermatites radio-induites et les érythèmes solaires peuvent réapparaître pendant le traitement par le méthotrexate (réaction de rappel). Les lésions psoriasiques peuvent s’aggraver en cas d’administration concomitante de méthotrexate pendant la PUVA thérapie.
L’élimination du méthotrexate est diminuée chez les patients présentant un espace de distribution tiers (ascite, épanchement pleural). Chez ces patients, il convient de surveiller étroitement les signes de toxicité et il peut être nécessaire de diminuer la posologie, ou, dans certains cas, d’arrêter le traitement par le méthotrexate. Les épanchements pleuraux et les ascites doivent être drainés avant l’instauration du traitement par le méthotrexate (voir rubrique 5.2).
Des diarrhées et une stomatite ulcérative peuvent être des effets toxiques et imposent l’interruption du traitement en raison du risque d’entérite hémorragique et de décès dû à une perforation intestinale.
Les préparations vitaminiques ou les autres produits contenant de l’acide folique, de l’acide folinique ou leurs dérivés peuvent diminuer l’efficacité du méthotrexate.
Dans le psoriasis, le traitement par le méthotrexate doit être limité aux formes modérées à sévères qui ne répondent pas suffisamment aux formes topiques de thérapie, mais uniquement lorsque le diagnostic a été confirmé par biopsie et/ou après une consultation dermatologique.
Des cas d’encéphalopathie/de leucoencéphalite ont été signalés chez des patients en oncologie recevant un traitement par le méthotrexate et la survenue de tels évènements ne peut pas être exclue dans le cas d’un traitement par le méthotrexate dans les indications non oncologiques.
Leucoencéphalopathie multifocale progressive (LEMP)
Des cas de leucoencéphalopathie multifocale progressive (LEMP) ont été rapportés chez des patients recevant du méthotrexate, le plus souvent en association avec d’autres médicaments immunosuppresseurs. La LEMP peut être fatale et doit être prise en compte dans le diagnostic différentiel chez les patients immunodéprimés présentant une nouvelle apparition ou une aggravation des symptômes neurologiques.
Fertilité et reproduction
Fertilité
Il a été constaté que le méthotrexate pouvait entraîner une oligospermie, un dysfonctionnement menstruel et une aménorrhée chez les humains, pendant le traitement et durant une brève période après l’arrêt de celui-ci, et une altération de la fertilité, affectant la spermatogenèse et l'ovogenèse durant la période de son administration ‑ effets qui semblent être réversibles après l'interruption du traitement.
Tératogénicité – risque inhérent à la reproduction
Le méthotrexate provoque une toxicité embryonnaire, des avortements et des malformations fœtales chez les humains. Par conséquent, les risques possibles d’effets sur la reproduction, de fausses couches et de malformations congénitales doivent être abordés avec les patientes fertiles (voir rubrique 4.6). L’absence de grossesse doit être confirmée avant d’utiliser METOJECT.
Si des femmes en âge de procréer sont traitées, une méthode de contraception efficace durant le traitement et pendant au moins six mois après l’arrêt du traitement doit être utilisée.
Pour des conseils sur les méthodes contraceptives masculines, voir rubrique 4.6.
METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu’il est essentiellement « sans sodium ».
Population pédiatrique
Ce médicament ne doit pas être utilisé chez l’enfant de moins de 3 ans compte tenu de l’insuffisance de données concernant la sécurité et l’efficacité du produit chez cette population (voir rubrique 4.2).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Protoxyde d’azote
L’utilisation de protoxyde d’azote potentialise l’effet du méthotrexate sur le métabolisme des folates, et conduit à une toxicité accrue telle qu’une myélosuppression sévère imprévisible et une stomatite. Bien qu’il soit possible d’atténuer cet effet par administration de folinate de calcium, l’utilisation concomitante de protoxyde d’azote et de méthotrexate doit être évitée.
Alcool, médicaments hépatotoxiques, médicaments hématotoxiques
Le risque d’effets hépatotoxiques du méthotrexate est majoré en cas de consommation régulière d’alcool ou d’administration concomitante d’autres médicaments hépatotoxiques (voir rubrique 4.4).
Une surveillance particulière s’impose chez les patients recevant simultanément d’autres médicaments hépatotoxiques (léflunomide par exemple). Les mêmes précautions doivent être prises en cas de co-administration de médicaments hématotoxiques (léflunomide, azathioprine, rétinoïdes, sulfasalazine par exemple). L’incidence de pancytopénie et l’hépatotoxicité peuvent être augmentées en cas d’association du léflunomide avec le méthotrexate.
Le risque d’hépatotoxicité est majoré en cas d’administration concomitante de méthotrexate et de rétinoïdes tels que l’acitrétine ou l’étrétinate.
Antibiotiques oraux
Les antibiotiques oraux tels que les tétracyclines, le chloramphénicol et les antibiotiques non absorbables à large spectre peuvent interférer avec la circulation entérohépatique par inhibition de la flore intestinale ou suppression du métabolisme bactérien.
Antibiotiques
Les antibiotiques de type pénicillines, glycopeptides, sulfamides, ciprofloxacine et céfalotine peuvent dans certains cas diminuer la clairance du méthotrexate, ce qui peut entraîner une augmentation des concentrations sériques du méthotrexate et une toxicité hématologique et gastro-intestinale.
Médicaments fortement liés aux protéines plasmatiques
Le méthotrexate se lie aux protéines plasmatiques et il peut être déplacé par les autres médicaments liés aux protéines tels que les salicylés, les hypoglycémiants, les diurétiques, les sulfamides, les diphénylhydantoïnes, les tétracyclines, le chloramphénicol et l’acide p-aminobenzoïque et les anti-inflammatoires acides, ce qui peut entraîner une toxicité accrue en cas d’administration concomitante.
Probénécide, acides organiques faibles, pyrazolés et anti-inflammatoires non stéroïdiens
Le probénécide, les acides organiques faibles tels que les diurétiques de l’anse et les pyrazolés (phénylbutazone) peuvent diminuer l’élimination du méthotrexate et des concentrations sériques plus élevées sont susceptibles de majorer la toxicité hématologique. La toxicité peut également être augmentée en cas d’association de méthotrexate à faible dose et d’anti-inflammatoires non stéroïdiens ou de salicylés.
Médicaments ayant des effets indésirables sur la moelle osseuse
Il convient de veiller au risque d’altération sévère de l’hématopoïèse en cas de traitement par des médicaments susceptibles d’avoir des effets indésirables sur la moelle osseuse (par exemple sulfamides, triméthoprime-sulfaméthoxazole, chloramphénicol, pyriméthamine). L’administration concomitante de métamizole et de méthotrexate peut augmenter l’effet hématotoxique du méthotrexate, en particulier chez les patients âgés. Par conséquent, l’administration concomitante doit être évitée.
Médicaments entraînant une carence en acide folique
L’administration concomitante de produits qui induisent une carence en acide folique (par exemple sulfamides, triméthoprime-sulfaméthoxazole) peut majorer la toxicité du méthotrexate. Une prudence particulière est donc recommandée en cas de déficit en acide folique préexistant.
Produits contenant de l’acide folique ou de l’acide folinique
Les préparations vitaminiques ou les autres produits contenant de l’acide folique, de l’acide folinique ou leurs dérivés peuvent diminuer l’efficacité du méthotrexate.
Autres antirhumatismaux
En général, il n’est pas attendu d’augmentation des effets toxiques du méthotrexate lorsque METOJECT est co-administré avec d’autres agents antirhumatismaux (par exemple sels d’or, pénicillamine, hydroxychloroquine, sulfasalazine, azathioprine).
Ciclosporine
La ciclosporine est susceptible de potentialiser l’efficacité et la toxicité du méthotrexate. Il en résulte un risque accru d’insuffisance rénale. En outre, il existe une possibilité biologique d’immunosuppression excessive et de complications associées.
Sulfasalazine
Bien que l’association de méthotrexate et de sulfasalazine puisse potentialiser l’efficacité du méthotrexate et par conséquent augmenter les effets indésirables dus à l’inhibition de la synthèse d’acide folique par la sulfasalazine, ces effets indésirables n’ont été observés que dans de rares cas isolés au cours de plusieurs études.
Mercaptopurine
Le méthotrexate augmente les concentrations plasmatiques de la mercaptopurine. Il peut donc être nécessaire d’adapter la posologie en cas d’association de méthotrexate et de mercaptopurine.
Inhibiteurs de la pompe à protons
L’administration concomitante d’inhibiteurs de la pompe à protons tels que l’oméprazole ou le pantoprazole peut provoquer des interactions : la co‑administration de méthotrexate et d’oméprazole a retardé l’élimination rénale du méthotrexate. L’association avec le pantoprazole a inhibé l’élimination rénale du métabolite 7‑hydroxyméthotrexate et des myalgies et des tremblements ont été observés chez un sujet.
Théophylline
Le méthotrexate peut diminuer la clairance de la théophylline ; les concentrations de théophylline doivent être surveillées en cas d’administration concomitante avec le méthotrexate.
Boissons contenant de la caféine ou de la théophylline
La consommation excessive de boissons contenant de la caféine ou de la théophylline (café, sodas contenant de la caféine, thé noir) doit être évitée pendant le traitement par le méthotrexate.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer/Contraception chez les femmes
Les femmes doivent éviter de tomber enceintes pendant le traitement par méthotrexate et une méthode de contraception efficace doit être utilisée pendant le traitement par méthotrexate et pendant au moins six mois après l’arrêt du traitement (voir rubrique 4.4). Avant le début du traitement, les femmes en âge de procréer doivent être informées du risque de malformations associées au méthotrexate et il convient d’exclure toute grossesse avec certitude en prenant des mesures appropriées, par exemple en réalisant un test de grossesse. Pendant le traitement, il convient de faire de nouveaux tests de grossesse si cela est jugé cliniquement pertinent (par exemple après une mauvaise observance de la contraception). Les patientes en âge de procréer doivent être conseillées sur la prévention et la planification d’une grossesse.
Contraception masculine
Il n'a pas été déterminé si le méthotrexate est présent dans le sperme. Les études chez l’animal ont mis en évidence une génotoxicité du méthotrexate, de sorte que le risque d’effets génotoxiques sur les spermatozoïdes ne peut pas être complètement exclu. Des preuves cliniques limitées n’indiquent pas un risque accru de malformations ou de fausses couches à la suite d’une exposition paternelle au méthotrexate à faible dose (moins de 30 mg/semaine). A plus fortes doses, les données sont insuffisantes pour estimer les risques de malformations ou de fausses couches à la suite d’une exposition paternelle.
Par mesure de précaution, il est recommandé aux patients de sexe masculin sexuellement actifs ou à leurs partenaires de sexe féminin d’utiliser une contraception fiable pendant le traitement du patient masculin et pendant au moins 3 mois après l'arrêt du traitement par méthotrexate. Les hommes ne doivent pas faire de don de sperme en cours de traitement ou pendant 3 mois après l'interruption du traitement par méthotrexate.
Grossesse
Les études chez l’animal ont mis en évidence une toxicité du méthotrexate sur la reproduction, notamment au cours du premier trimestre (voir rubrique 5.3). Le méthotrexate s’est avéré tératogène chez les humains ; des cas de mort fœtale, des fausses couches et/ou des anomalies congénitales ont été rapportés (par exemple, cranio-faciales, cardiovasculaires, du système nerveux central et des extrémités).
Le méthotrexate est un puissant « tératogène » humain, associé à un risque accru d'avortements spontanés, de retard de croissance intra-utérine et de malformations congénitales en cas d’exposition pendant la grossesse.
· Des avortements spontanés ont été rapportés chez 42,5 % des femmes enceintes exposées à un traitement par méthotrexate à faible dose (moins de 30 mg/semaine), contre un taux de 22,5 % rapporté chez des patientes dont la maladie présente des caractéristiques comparables et qui sont traitées par des médicaments autres que le méthotrexate.
· Des anomalies congénitales majeures ont été observées pour 6,6 % des naissances vivantes chez les femmes exposées à un traitement par méthotrexate à faible dose (moins de 30 mg/semaine) pendant la grossesse, contre approximativement 4 % des naissances vivantes chez des patientes dont la maladie présente des caractéristiques comparables et qui sont traitées par des médicaments autres que le méthotrexate.
L’exposition pendant la grossesse à des doses de méthotrexate supérieures à 30 mg/semaine est insuffisamment documentée, mais des taux plus élevés d’avortements spontanés et de malformations congénitales sont attendus.
Des cas de grossesses normales ont été rapportés lorsque le méthotrexate était arrêté avant la conception.
Allaitement
Le méthotrexate est excrété dans le lait maternel. Compte tenu du risque de réactions indésirables graves pour le nourrisson allaité, METOJECT est contre-indiqué pendant l’allaitement (voir rubrique 4.3). L’allaitement doit par conséquent être interrompu avant et pendant le traitement.
Fertilité
Le méthotrexate affecte la spermatogenèse et l'ovogenèse et peut diminuer la fertilité. Chez les humains, il a été constaté que le méthotrexate pouvait entraîner une oligospermie, un dysfonctionnement menstruel et une aménorrhée. Dans la plupart des cas, ces effets semblent être réversibles après l'interruption du traitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Des symptômes nerveux centraux, tels que fatigue et étourdissements, peuvent survenir pendant le traitement.
Résumé du profil de sécurité
La plupart des effets indésirables graves du méthotrexate comprennent : aplasie médullaire, toxicité pulmonaire, hépatotoxicité, toxicité rénale, neurotoxicité, accidents thromboemboliques, choc anaphylactique et syndrome de Stevens-Johnson.
Les effets indésirables du méthotrexate ayant été observés le plus fréquemment (très fréquents) comprennent des affections gastro-intestinales (p. ex. stomatite, dyspepsie, douleurs abdominales, nausées, perte d'appétit) et des anomalies des tests de la fonction hépatique (p. ex. élévation de l’ALAT, de l’ASAT, de la bilirubine et de la phosphatase alcaline). Les autres effets indésirables qui surviennent fréquemment (fréquents) sont la leucopénie, l’anémie, la thrombopénie, les céphalées, la fatigue, la somnolence, la pneumonie, l’alvéolite/la pneumonie interstitielle souvent associée à une éosinophilie, les ulcérations buccales, la diarrhée, l’exanthème, l’érythème et le prurit.
Liste des effets indésirables
Les effets indésirables les plus significatifs sont une inhibition du système hématopoïétique et des troubles gastro-intestinaux.
Les effets indésirables sont présentés par ordre de fréquence selon la convention suivante :
Très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Infections et infestations
Peu fréquent : pharyngite.
Rare : infection (y compris réactivation d’une infection chronique inactive), sepsis, conjonctivite.
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)
Très rare : lymphome (voir la « description » ci-après).
Affections hématologiques et du système lymphatique
Fréquent : leucopénie, anémie, thrombopénie.
Peu fréquent : pancytopénie.
Très rare : agranulocytose, aplasie médullaire d’évolution sévère, syndrome lymphoprolifératif (voir la « description » ci-après).
Fréquence indéterminée : éosinophilie.
Affections du système immunitaire
Rare : réactions allergiques, choc anaphylactique, hypogammaglobulinémie.
Troubles du métabolisme et de la nutrition
Peu fréquent : apparition du diabète.
Affections psychiatriques
Peu fréquent : dépression, confusion.
Rare : altérations de l’humeur.
Affections du système nerveux
Fréquent : céphalées, fatigue, somnolence.
Peu fréquent : étourdissements.
Très rare : algies, asthénie musculaire ou paresthésie/hypoesthésie, dysgueusie (goût métallique), convulsions, méningisme, méningite aseptique aiguë, paralysie.
Fréquence indéterminée : encéphalopathie/leucoencéphalite.
Affections oculaires
Rare : troubles visuels, qui peut être associées à la neuropathie optique.
Très rare : baisse de la vision, rétinopathie.
Affections cardiaques
Rare : péricardite, épanchement péricardique, tamponnade péricardique.
Affections vasculaires
Rare : hypotension, accidents thromboemboliques.
Affections respiratoires, thoraciques et médiastinales
Fréquent : pneumonie, alvéolite/pneumonie interstitielle souvent associée à une éosinophilie. Les symptômes indiquant une atteinte pulmonaire potentiellement sévère (pneumonie interstitielle) sont : toux sèche non productive, essoufflement et fièvre.
Rare : fibrose pulmonaire, pneumonie à Pneumocystis jirovecii, essoufflement et asthme bronchique, épanchement pleural.
Fréquence indéterminée : épistaxis, hémorragie alvéolaire pulmonaire.
Affections gastro-intestinales
Très fréquent : stomatite, dyspepsie, nausées, perte d’appétit, douleurs abdominales.
Fréquent : ulcérations buccales, diarrhées.
Peu fréquent : ulcères et saignements gastro-intestinaux, entérite, vomissements, pancréatite.
Rare : gingivite.
Très rare : hématémèse, hémorragies abondantes, colectasie.
Affections hépatobiliaires (voir rubrique 4.4)
Très fréquent : anomalies des tests de la fonction hépatique (élévation de l’ALAT, de l’ASAT, de la phosphatase alcaline et de la bilirubine).
Peu fréquent : cirrhose, fibrose et stéatose hépatique, hypoalbuminémie.
Rare : hépatite aiguë.
Très rare : insuffisance hépatique.
Affections de la peau et du tissu sous-cutané
Fréquent : exanthème, érythème, prurit.
Peu fréquent : réactions de photosensibilité, alopécie, augmentation des nodosités rhumatismales, ulcères cutanés, zona, vascularite, éruptions cutanées herpétiformes, urticaire.
Rare : hyperpigmentation, acné, pétéchies, ecchymoses, vascularite allergique.
Très rare : syndrome de Stevens-Johnson, nécro-épidermolyse bulleuse aiguë (syndrome de Lyell), hyperpigmentation des ongles, panaris aigu, furonculose, télangiectasies.
Fréquence indéterminée : exfoliation cutanée/dermatite exfoliative.
Affections musculosquelettiques et du tissu conjonctif
Peu fréquent : arthralgies, myalgies, ostéoporose.
Rare : fracture de fatigue.
Fréquence indéterminée : ostéonécrose de la mâchoire (secondaire au syndrome lymphoprolifératif).
Affections du rein et des voies urinaires
Peu fréquent : inflammation et ulcération de la vessie, diminution de la fonction rénale, troubles de la miction.
Rare : insuffisance rénale, oligurie, anurie, déséquilibres électrolytiques.
Fréquence indéterminée : protéinurie.
Affections des organes de reproduction et du sein
Peu fréquent : inflammation et ulcération vaginales.
Très rare : diminution de la libido, impuissance, gynécomastie, oligospermie, aménorrhées, pertes vaginales.
Troubles généraux et anomalies au site d'administration
Rare : fièvre, difficultés de cicatrisation.
Fréquence indéterminée : asthénie, nécrose au point d’injection, œdème.
Description de certains effets indésirables
La survenue et la sévérité des effets indésirables dépendent de la dose utilisée et de la fréquence d’administration. Cependant, des effets indésirables sévères pouvant même survenir à faibles doses, une surveillance régulière et fréquente des patients s’avère indispensable.
Lymphome/syndrome lymphoprolifératif : des cas particuliers de lymphomes et d’autres syndromes lymphoprolifératifs, qui ont diminué dans un certain nombre de cas après l’interruption du traitement par méthotrexate, ont été rapportés.
L’administration sous-cutanée du méthotrexate est bien tolérée localement. Seules des réactions cutanées locales bénignes (telles que sensations de cuisson, érythème, œdème, décoloration, prurit, démangeaisons sévères et douleurs) régressant pendant le traitement, ont été observées.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Symptômes d’un surdosage
La toxicité du méthotrexate affecte essentiellement le système hématopoïétique.
Conduite à tenir en cas de surdosage
Le folinate de calcium est l’antidote spécifique pour neutraliser les effets toxiques du méthotrexate.
En cas de surdosage accidentel, une dose de folinate de calcium égale ou supérieure à la dose de méthotrexate reçue par le patient doit être administrée par voie intraveineuse ou intramusculaire dans un délai d'une heure, et l'administration doit se poursuivre jusqu'à ce que la concentration sérique de méthotrexate soit inférieure à 10‑7 mol/l.
En cas de surdosage massif, une hydratation et une alcalinisation des urines peuvent être nécessaires pour empêcher la précipitation du méthotrexate et/ou de ses métabolites dans les tubules rénaux. Ni l’hémodialyse, ni la dialyse péritonéale ne se sont révélées efficaces pour améliorer l’élimination du méthotrexate. Une clairance efficace du méthotrexate a été observée en cas d’hémodialyse immédiate intermittente à l’aide d’un dialyseur à haut débit.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : autres immunosuppresseurs, code ATC : L04AX03.
Médicament antirhumatismal destiné au traitement des maladies rhumatismales inflammatoires chroniques et des formes polyarticulaires de l’arthrite juvénile idiopathique. Agent immunomodulateur et anti-inflammatoire destiné au traitement de la maladie de Crohn.
Mécanisme d’action
Le méthotrexate est un antagoniste de l’acide folique qui appartient à la classe d’agents cytotoxiques appelés antimétabolites. Il agit par inhibition compétitive de l'enzyme dihydrofolate réductase et inhibe ainsi la synthèse de l’ADN.
Il n’a pas encore été clairement déterminé si l’efficacité du méthotrexate dans le traitement du psoriasis, du rhumatisme psoriasique, de la polyarthrite chronique et de la maladie de Crohn est due à un effet anti-inflammatoire ou immunodépresseur et dans quelle mesure l’augmentation induite par le méthotrexate de la concentration d’adénosine extracellulaire dans les sites inflammatoires contribue à ces effets.
Les recommandations cliniques internationales font état de l’utilisation du méthotrexate en deuxième intention chez les patients atteints de la maladie de Crohn qui ont présenté une intolérance ou un échec thérapeutique lors du traitement de première intention par des agents immunomodulateurs tels que l’azathioprine (AZA) ou la 6‑mercaptopurine (6‑MP).
Les effets indésirables observés lors des études menées avec le méthotrexate pour le traitement de la maladie de Crohn en administration répétée n’ont pas révélé un profil de sécurité du méthotrexate différent du profil déjà connu. Par conséquent, lors de l’utilisation du méthotrexate pour le traitement de la maladie de Crohn, les mêmes précautions doivent être prises que pour les autres indications du méthotrexate dans le traitement de maladies rhumatismales et non rhumatismales (voir les rubriques 4.4 et 4.6).
Efficacité clinique et tolérance
Un essai en double aveugle, multicentrique, randomisé, clinique de phase IV (MC-MTX.6/RA) a été mené pour comparer l'efficacité et la tolérance de MTX par voie sous-cutanée (SC) par rapport au MTX par voie orale chez des patients adultes naïfs de MTX atteints de polyarthrite rhumatoïde active (DAS28 ≥ 4). Les patients ont reçu une dose de 15 mg/semaine de MTX par voie orale (2 comprimés de MTX plus 1 injection par voie SC placebo, n = 190) ou par voie SC (15 mg par voie SC de MTX plus 2 comprimés placebo, n = 194) pendant 24 semaines. A la semaine 16, les patients qui n'ont pas eu de réponse ACR20 sont passés de 15 mg de MTX par voie orale à 15 mg de MTX par voie SC et de 15 mg de MTX par voie SC à 20 mg par voie SC pendant les 8 semaines restantes. Le critère d’évaluation principal était le pourcentage de patients répondeurs ACR20 à la semaine 24. Les critères d’évaluation secondaires incluaient les réponses ACR50 et ACR70 ainsi que la tolérance au MTX par voie SC par rapport à une administration par voie orale. Dans le groupe voie SC, 78,2 % des patients étaient répondeurs ACR20 à la semaine 24 par rapport à 70,1 % dans le groupe voie orale (P = 0,0412). Dans le groupe voie SC, significativement plus de patients étaient répondeurs ACR70 comparé au groupe voie orale à la semaine 24 (41,0 % contre 33,2 %, P = 0,03). L'incidence globale des effets indésirables (EI), au cours de la durée totale de l'essai, dans la population analysée pour la tolérance, a été similaire dans les deux groupes de traitement, avec des EI signalés chez 66,3 % des patients du groupe voie SC et 61,7 % du groupe voie orale. L'incidence globale des effets indésirables graves (EIG) était également similaire dans les deux groupes traités (5,7 % versus 4,3 %).
Une autre étude multicentrique a comparé la satisfaction des patients et la tolérance locale aux injections SC répétées de MTX 50 mg/mL ou 10 mg/mL chez des patients atteints de polyarthrite rhumatoïde active. Au total, 132 patients étaient inscrits dans 16 centres investigateurs en Allemagne. 93 % des participants ont déclaré qu'ils préféraient la forme la plus concentrée de MTX à 50 mg/mL (P <0,0001). Des effets indésirables liés au médicament ont été signalés chez 24 patients (18,3 %). La concentration de MTX à 50 mg/mL était légèrement mieux tolérée tant par l'évaluation du médecin que par celle du patient.
L'essai en double aveugle, contrôlé versus placebo, de phase III « METOP » a été mené chez 120 patients adultes naïfs de MTX et atteints de psoriasis en plaques chronique modéré à sévère diagnostiqué depuis au moins 6 mois. Les patients inclus ont été répartis de manière aléatoire (3:1) pour recevoir soit du MTX à une dose initiale de 17,5 mg/semaine (n = 91), soit le placebo (n = 29) pendant les 16 premières semaines. Par la suite, tous les patients ont été traités par MTX par voie SC jusqu'à la semaine 52 (groupes MTX-MTX versus placebo-MTX). L’augmentation de la dose à 22,5 mg/semaine était autorisée après 8 semaines de traitement par MTX si les patients n'avaient pas atteint une réduction d'au moins 50 % du score PASI (Psoriasis Areas Severity Index) initial, la même augmentation du volume injecté a été également appliquée aux injections de placebo. Le traitement a été associé à l’administration d’acide folique, à une posologie de 5 mg/semaine. A la semaine 16, une réponse PASI 75 a été atteinte chez 37 (41 %) patients du groupe MTX contre trois (10 %) patients dans le groupe placebo (RR 3,93 ; P = 0,0026). Le MTX administré par voie SC a été généralement bien toléré ; aucun cas de décès n’a été noté, aucun patient n’a souffert d’infections graves, de tumeurs malignes ou d’événements cardiovasculaires majeurs. Des événements indésirables graves ont été répertoriés chez trois patients (3 %) ayant reçu du MTX pendant toute la période de traitement de 52 semaines.
5.2. Propriétés pharmacocinétiques
Après administration orale, le méthotrexate est absorbé au niveau des voies gastro-intestinales. En cas d’administration à faibles doses (doses de 7,5 mg/m2 à 80 mg/m2 de surface corporelle), la biodisponibilité moyenne est d’environ 70 %, mais des variations interindividuelles et intra-individuelles importantes sont possibles (de 25 à 100 %). Le pic de concentration sérique est atteint en 1 à 2 heures.
La biodisponibilité est comparable et proche des 100 % en cas d’administration sous-cutanée, intraveineuse et intramusculaire.
Distribution
La liaison du méthotrexate aux protéines sériques est d’environ 50 %. Après distribution dans les tissus corporels, des concentrations élevées sous forme de polyglutamates sont observées notamment dans le foie, les reins et la rate et elles peuvent persister pendant des semaines, voire des mois. Lorsqu’il est administré à faibles doses, le méthotrexate passe dans le liquide cérébro-spinal en quantités minimes. La demi-vie terminale est de 6 à 7 heures en moyenne, avec des variations considérables (3 à 17 heures). La demi-vie du méthotrexate peut être prolongée jusqu’à 4 fois la valeur normale chez les patients qui présentent un espace de distribution tiers (épanchement pleural, ascite).
Biotransformation
Environ 10 % de la dose de méthotrexate administrée sont métabolisés dans le foie. Le principal métabolite est le 7‑hydroxyméthotrexate.
Élimination
Le méthotrexate est éliminé essentiellement sous forme inchangée, par voie rénale principalement, par filtration glomérulaire et sécrétion active dans les tubules proximaux.
Environ 5 % à 20 % du méthotrexate et 1 % à 5 % du 7‑hydroxyméthotrexate sont éliminés par voie biliaire. La circulation entérohépatique est importante.
L’élimination est fortement retardée en cas de troubles de la fonction rénale. Les effets d’une altération de la fonction hépatique sur l’élimination ne sont pas connus.
5.3. Données de sécurité préclinique
Hydroxyde de sodium (pour l’ajustement du pH) ;
Acide chlorhydrique (pour l’ajustement du pH) ;
Eau pour préparations injectables.
30 mois.
6.4. Précautions particulières de conservation
À conserver à une température ne dépassant pas 25 °C. Ne pas congeler.
Conserver les stylos préremplis dans l’emballage extérieur, à l’abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
Stylos préremplis contenant une seringue préremplie (en verre incolore) avec bouchon piston (caoutchouc chlorobutyle) et aiguille d’injection SC fixée. La seringue est intégrée dans un dispositif (stylo) permettant une auto-administration du produit.
Le stylo prérempli METOJECT est disponible sous la forme d’un auto‑injecteur en trois étapes, muni d’un capuchon jaune et d’un bouton poussoir jaune ou d’un auto‑injecteur en deux étapes, muni d’un capuchon de protection translucide et d’un protège‑aiguille bleu.
Présentations :
Stylos préremplis contenant 0,35 ml (17,5 mg) de solution présentés en boîtes de 1, 2, 4, 5, 6, 10, 11, 12, 14, 15 et 24 stylos préremplis.
Toutes les présentations peuvent ne pas être commercialisées.
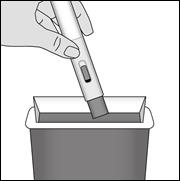
6.6. Précautions particulières d’élimination et de manipulation
Les procédures pour la manipulation et l’élimination doivent respecter les exigences et réglementations locales. Les femmes enceintes appartenant au personnel soignant ne doivent pas manipuler et/ou administrer METOJECT.
Le méthotrexate ne doit pas entrer en contact avec la peau ou les muqueuses. En cas de contamination, la région affectée doit être rincée immédiatement et abondamment à l’eau.
À usage unique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
MEDAC GESELLSCHAFT FÜR KLINISCHE SPEZIALPRÄPARATE MBH
THEATERSTR. 6
22880 WEDEL
ALLEMAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 300 203 7 1 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 2
· 34009 300 203 8 8 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 4
· 34009 550 070 8 1 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 5
· 34009 550 070 9 8 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 6
· 34009 550 071 0 4 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 10
· 34009 550 071 1 1 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 11
· 34009 550 071 2 8 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 12
· 34009 550 071 3 5 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 14
· 34009 550 071 4 2 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 15
· 34009 550 071 5 9 : 0,35 ml de solution injectable en stylo prérempli (verre) avec aiguille d’injection SC fixée ; boîte de 24
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 15/10/2024
METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli
Méthotrexate
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli ?
3. Comment utiliser METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique – code ATC : autres immunosuppresseurs - L04AX03.
METOJECT est indiqué dans le traitement :
· de la polyarthrite rhumatoïde sévère et active chez l’adulte ;
· des formes polyarticulaires de l’arthrite juvénile idiopathique active sévère, en cas de réponse inadéquate aux anti‑inflammatoires non stéroïdiens (AINS) ;
· des formes modérées à sévères du psoriasis chez les patients adultes et des formes sévères du rhumatisme psoriasique chez l’adulte ;
· des formes légères à modérées de la maladie de Crohn chez l’adulte lorsqu’un traitement adéquat par d’autres médicaments n’est pas possible.
La polyarthrite rhumatoïde (PR) est une maladie chronique du collagène caractérisée par une inflammation des membranes synoviales (membranes des articulations). Ces membranes sécrètent un liquide qui agit comme un lubrifiant pour de nombreuses articulations. L’inflammation provoque un épaississement de la membrane et un gonflement de l’articulation.
L’arthrite juvénile est une maladie qui touche les enfants et adolescents de moins de 16 ans. On parle de formes polyarticulaires lorsque 5 articulations ou plus sont touchées au cours des six premiers mois de la maladie.
Le psoriasis est une affection cutanée chronique courante caractérisée par des plaques rouges recouvertes de squames adhérentes nacrées, épaisses et sèches.
Le rhumatisme psoriasique est une forme d’arthrite accompagnée de lésions psoriasiques de la peau et des ongles, notamment au niveau des articulations des doigts et des orteils.
METOJECT modifie et ralentit la progression de la maladie.
La maladie de Crohn est un type de maladie inflammatoire des intestins qui peut affecter n’importe quelle région du tube gastro-intestinal et provoque des symptômes tels que des douleurs abdominales, de la diarrhée, des vomissements ou une perte de poids.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli ?
N’utilisez jamais METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli :
· si vous êtes allergique au méthotrexate ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6 ;
· si vous souffrez d’une maladie du foie ou d’une maladie des reins sévère ou d’une affection du sang ;
· si vous consommez régulièrement des quantités importantes d’alcool ;
· si vous avez une infection grave, comme la tuberculose, une infection par le VIH ou d’autres syndromes d’immunodéficience ;
· si vous avez des ulcérations buccales, un ulcère de l’estomac ou un ulcère intestinal ;
· si vous êtes enceinte ou si vous allaitez (voir la rubrique « Grossesse, allaitement et fertilité ») ;
· si vous devez recevoir en même temps des vaccinations avec des vaccins vivants.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser METOJECT si :
· vous êtes une personne âgée ou si vous vous sentez généralement souffrant(e) et faible ;
· vous avez des troubles de la fonction hépatique ;
· vous souffrez de déshydratation (manque d’eau) ;
· vous êtes diabétique et êtes traité par de l’insuline.
Mesures de précaution spéciales pour le traitement par METOJECT
Le méthotrexate affecte temporairement la production du sperme et des ovules, ces effets étant réversibles dans la plupart des cas. Le méthotrexate peut provoquer des fausses couches et de graves anomalies congénitales. Si vous êtes une femme, vous devez éviter de tomber enceinte pendant que vous prenez du méthotrexate et pendant au moins 6 mois après l’arrêt du traitement. Si vous êtes un homme, vous devez éviter de concevoir un enfant pendant que vous prenez du méthotrexate et pendant au moins 3 mois après l’arrêt du traitement. Voir également la rubrique « Grossesse, allaitement et fertilité ».
Examens de contrôle recommandés et précautions
Même si le méthotrexate est administré à faibles doses, des effets indésirables graves peuvent survenir. Afin de les détecter à temps, votre médecin devra pratiquer des examens de contrôle et demander des analyses biologiques.
Avant le début du traitement
Avant le début du traitement, des analyses de sang seront effectuées pour vérifier que vous avez suffisamment de cellules sanguines. Des analyses de sang seront également effectuées pour contrôler votre fonction hépatique et pour un dépistage de l’hépatite. De plus, votre taux d’albumine sérique (une protéine présente dans le sang), votre statut pour l’hépatite (infection du foie) et votre fonction rénale seront contrôlés. Le médecin pourra également décider de réaliser d’autres examens du foie, certains pouvant être des examens d’imagerie et d’autres pouvant nécessiter le prélèvement d’un petit échantillon de tissu hépatique afin qu’il soit examiné de plus près. Votre médecin pourra également vérifier si vous avez la tuberculose et pourra demander une radiographie pulmonaire ou réaliser des tests de la fonction pulmonaire.
Pendant le traitement
Votre médecin pourra effectuer ou demander les examens suivants :
· examen de la cavité buccale et du pharynx pour détecter des modifications des muqueuses telles qu’une inflammation ou des ulcérations ;
· analyses de sang/numération la formule sanguine et dosage des concentrations sériques de méthotrexate ;
· analyse de sang pour contrôler la fonction hépatique ;
· examens d’imagerie pour contrôler l’état du foie ;
· prélèvement d’un petit échantillon de tissu hépatique afin qu’il soit examiné de plus près ;
· analyse de sang pour contrôler la fonction rénale ;
· contrôle des voies respiratoires et, si nécessaire, tests de la fonction pulmonaire.
Il est très important que vous respectiez les rendez-vous pour ces examens programmés.
Si les résultats de l’un de ces examens sont anormaux, votre médecin ajustera votre traitement en conséquence.
Patients âgés
Les patients âgés traités par le méthotrexate doivent être étroitement surveillés par un médecin afin que les éventuels effets indésirables puissent être détectés le plus tôt possible.
La diminution des fonctions hépatique et rénale liée à l’âge ainsi que les faibles réserves corporelles en acide folique (une vitamine) à un âge avancé nécessitent une dose relativement faible de méthotrexate.
Autres précautions
Des saignements aigus au niveau des poumons chez des patients souffrant de pathologies rhumatologiques sous-jacentes ont été rapportés lors de traitements par méthotrexate. Si vous présentez des symptômes tels qu'expectorations (crachats) ou toux accompagnées de sang, contactez immédiatement votre médecin.
Le méthotrexate peut affecter votre système immunitaire et votre réponse aux vaccinations. Il peut également modifier les résultats des tests immunologiques. Des infections chroniques inactives (comme le zona, la tuberculose, l’hépatite B ou C) peuvent se réactiver. Vous ne devez pas recevoir de vaccins vivants pendant le traitement par METOJECT.
Le méthotrexate peut rendre votre peau plus sensible au soleil. Évitez de vous exposer au soleil intense et n’utilisez pas de solarium ou de lampe à ultra-violets sans avis médical. Pour protéger votre peau du soleil intense, portez des vêtements adéquats ou utilisez une crème solaire à indice de protection élevé.
Les dermatites radio-induites et les érythèmes solaires peuvent réapparaître pendant le traitement par le méthotrexate (réaction de rappel). Les lésions psoriasiques peuvent s’aggraver en cas d’administration simultanée de méthotrexate pendant un traitement par rayons UV.
Une hypertrophie des ganglions lymphatiques (lymphome) peut survenir ; dans ce cas, le traitement doit être arrêté.
La diarrhée est un effet indésirable potentiel de METOJECT nécessitant une interruption du traitement. En cas de diarrhées, consultez votre médecin.
Certains troubles cérébraux (encéphalopathie/leucoencéphalite) ont été signalés chez des patients atteints de cancer recevant un traitement par le méthotrexate. La survenue de tels effets indésirables ne peut pas être exclue lorsque le méthotrexate est utilisé pour traiter d’autres maladies.
Si vous, votre partenaire ou votre aidant remarquez une nouvelle apparition ou une aggravation de symptômes neurologiques, notamment une faiblesse musculaire générale, des troubles de la vision, des changements de pensée, de mémoire et d’orientation entraînant une confusion et des modifications de la personnalité, contactez immédiatement votre médecin car il peut s’agir de symptômes d’une infection cérébrale grave très rare appelée leucoencéphalopathie multifocale progressive (LEMP).
Enfants et adolescents
Sans objet.
Autres médicaments et METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament. Ceci concerne également les médicaments que vous pourriez prendre dans un avenir plus ou moins proche.
Les effets du traitement peuvent être modifiés si METOJECT est administré en même temps que certains autres médicaments :
· les antibiotiques tels que : tétracyclines, chloramphénicol et antibiotiques non absorbables à large spectre, pénicillines, glycopeptides, sulfamides, ciprofloxacine et céfalotine (médicaments utilisés pour prévenir/combattre certaines infections) ;
· les anti-inflammatoires non stéroïdiens ou les salicylés (médicaments utilisés pour soulager la douleur et/ou l’inflammation tels que l’acide acétylsalicylique, le diclofénac et l’ibuprofène, ou les pyrazolés) ;
· le métamizole (synonymes novaminsulfone et dipyrone) (médicaments contre les fortes douleurs et/ou la fièvre) ;
· le probénécide (médicament utilisé dans le traitement de la goutte) ;
· les acides organiques faibles comme les diurétiques de l’anse (médicaments augmentant la production d’urine) ;
· les médicaments qui peuvent avoir des effets indésirables sur la moelle osseuse, comme le triméthoprime-sulfaméthoxazole (un antibiotique) et la pyriméthamine ;
· d’autres médicaments utilisés pour traiter la polyarthrite rhumatoïde tels que le léflunomide, la sulfasalazine et l’azathioprine ;
· la ciclosporine (pour la suppression du système immunitaire) ;
· la mercaptopurine (un médicament cytostatique) ;
· les rétinoïdes (médicaments utilisés dans le traitement du psoriasis et d’autres maladies de la peau) ;
· la théophylline (médicament utilisé pour traiter l’asthme et d’autres maladies pulmonaires) ;
· certains médicaments utilisés pour soulager les troubles gastriques tels que l’oméprazole et le pantoprazole ;
· les hypoglycémiants (médicaments utilisés pour faire diminuer le taux de sucre dans le sang) ;
· les préparations vitaminiques contenant de l’acide folique peuvent modifier l’effet de votre traitement et vous ne devez les prendre qu’avec l’accord de votre médecin.
La vaccination avec des vaccins vivants doit impérativement être évitée pendant le traitement.
METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli avec des aliments, boissons et de l’alcool
L’alcool et la consommation en quantités importantes de café, de sodas contenant de la caféine et de thé noir doivent être évités pendant le traitement par METOJECT.
Grossesse, allaitement et fertilité
Grossesse
N’utilisez pas METOJECT pendant la grossesse ou si vous essayez de tomber enceinte. Le méthotrexate peut provoquer des anomalies congénitales, avoir des effets nocifs sur l'enfant à naître ou provoquer des fausses couches. Il est associé à des malformations du crâne, du visage, du cœur et des vaisseaux sanguins, du cerveau et des membres. Il est, dès lors, extrêmement important de ne pas administrer de méthotrexate aux patientes enceintes ou à celles qui envisagent de le devenir. Chez les femmes en âge de procréer, toute possibilité de grossesse doit être exclue en prenant des mesures appropriées, par exemple en réalisant un test de grossesse avant le début du traitement. Vous devez éviter de tomber enceinte pendant la prise de méthotrexate et pendant au moins 6 mois après l’arrêt du traitement en utilisant une contraception fiable durant toute cette période (voir également la rubrique « Avertissements et précautions »).
Si vous tombez enceinte au cours du traitement ou si vous pensez l’être, consultez votre médecin dès que possible. Une information médicale sur les risques d'effets nocifs du méthotrexate sur l’enfant doit vous être fournie.
Si vous souhaitez tomber enceinte, vous devez consulter votre médecin, qui pourra vous adresser à un spécialiste avant le début planifié du traitement.
Allaitement
L’allaitement doit être interrompu avant et pendant le traitement par METOJECT.
Fertilité masculine
Les preuves disponibles n’indiquent pas d’augmentation du risque de malformations ou de fausses couches si le père prend moins de 30 mg/semaine de méthotrexate. Un risque ne peut toutefois pas être complètement exclu. Le méthotrexate peut être génotoxique. Cela signifie que le médicament peut provoquer des mutations génétiques. Le méthotrexate peut affecter la production du sperme, ce qui peut provoquer des anomalies congénitales. Par conséquent, vous devriez éviter de concevoir un enfant ou de faire un don de sperme pendant la prise de méthotrexate et pendant au moins 3 mois après l’arrêt du traitement.
Conduite de véhicules et utilisation de machines
Le traitement par METOJECT peut entraîner des effets indésirables sur le système nerveux central, comme une fatigue ou des étourdissements. Par conséquent, la capacité à conduire un véhicule et/ou à utiliser des machines peut être altérée dans certains cas. Si vous vous sentez fatigué(e) ou somnolent(e), vous ne devez pas conduire ou utiliser de machines.
METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli ?
Avertissement important concernant la dose de METOJECT 17,5 mg / 0,35 ml (méthotrexate) :
Utilisez METOJECT 17,5 mg / 0,35 ml une fois par semaine seulement pour le traitement de la polyarthrite rhumatoïde, de l’arthrite juvénile idiopathique, du psoriasis, du rhumatisme psoriasique et de la maladie de Crohn. L’utilisation d’une dose excessive de METOJECT 17,5 mg / 0,35 ml (méthotrexate) peut avoir des conséquences fatales. Veuillez lire très attentivement la rubrique 3 de cette notice. Si vous avez des questions, veuillez-vous adresser à votre médecin ou à votre pharmacien avant de prendre ce médicament.
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Votre médecin déterminera la posologie qui est adaptée personnellement à chaque patient. En général, l’effet du traitement apparaît après 4 à 8 semaines.
Seule la première injection de METOJECT doit être administrée par ou sous la surveillance d’un médecin ou d’un professionnel de la santé. METOJECT est injecté une fois par semaine seulement. Vous déciderez avec votre médecin du jour de la semaine approprié pour recevoir votre injection hebdomadaire.
Utilisation chez les enfants et les adolescents
Chez l’enfant et l’adolescent atteint de la forme polyarticulaire de l’arthrite juvénile idiopathique, la dose appropriée sera déterminée par le médecin. METOJECT ne doit pas être utilisé chez l’enfant de moins de 3 ans compte tenu de l’expérience insuffisante dans ce groupe d’âge.
Mode d’administration et durée du traitement
METOJECT est injecté une fois par semaine.
La durée du traitement est déterminée par le médecin traitant. Le traitement de la polyarthrite rhumatoïde, de l’arthrite juvénile idiopathique, du psoriasis en plaques, du rhumatisme psoriasique et de la maladie de Crohn par METOJECT est un traitement de longue durée.
Au début de votre traitement, METOJECT peut vous être injecté par du personnel médical. Toutefois, votre médecin peut décider que vous pouvez apprendre à vous injecter METOJECT vous-même. Vous serez alors convenablement formé à l’auto-injection. En aucun cas vous ne devrez essayer de vous auto-injecter le produit si vous n’avez pas été formé à cet effet.
Après une formation appropriée, vous pourrez administrer l’injection vous-même, ou elle pourra être administrée par une autre personne, par exemple un membre de votre famille ou un ami.
Vous ne devez sous aucun prétexte essayer de vous injecter vous-même METOJECT avant d’avoir suivi cette formation.
Veuillez lire le mode d’emploi à la fin de cette notice pour trouver des conseils sur la manière d’utiliser correctement METOJECT.
Notez que l’intégralité du produit contenu dans le stylo doit être utilisée à chaque injection.
Les procédures pour la manipulation et l’élimination de ce produit doivent respecter les exigences et réglementations locales. Les femmes enceintes appartenant au personnel soignant ne doivent pas manipuler et/ou administrer METOJECT.
Le méthotrexate ne doit pas entrer en contact avec la peau ou les muqueuses. En cas de contact, la région affectée doit être rincée immédiatement et abondamment à l’eau.
Si vous avez utilisé plus de METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli que vous n’auriez dû
Si vous avez utilisé trop de METOJECT, prenez immédiatement contact avec votre médecin.
Si vous oubliez d’utiliser METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez d’utiliser METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli
Si vous arrêtez d’utiliser METOJECT, parlez-en à votre médecin immédiatement.
Si vous avez l’impression que l’effet de METOJECT est trop fort ou trop faible, consultez votre médecin ou votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
La fréquence et le niveau de gravité des effets indésirables dépendent de la dose utilisée et de la fréquence d’administration. Comme des effets indésirables sévères peuvent survenir même à faibles doses, il est important que vous soyez suivi(e) régulièrement par votre médecin. Votre médecin procédera alors à des analyses visant à vérifier l’absence d’anomalies sanguines (comme une chute du nombre des globules blancs, des plaquettes ou la présence d’un lymphome) et de modifications d’origine rénale ou hépatique.
Prenez immédiatement contact avec votre médecin si vous ressentez l’un des symptômes suivants, car ceux-ci peuvent révéler l’apparition d’un effet secondaire grave, pouvant mettre en jeu la vie du patient, et nécessitant la mise en place d’un traitement spécifique d’urgence :
· une toux sèche, non productive et persistante, un essoufflement et de la fièvre, ces signes pouvant être le signe d’une inflammation des poumons [fréquent] ;
· des expectorations (crachats) ou des toux accompagnées de sang, ceci peut être le signe de saignements au niveau des poumons [fréquence indéterminée] ;
· des symptômes d’une atteinte hépatique, comme une coloration jaune de la peau et du blanc des yeux, car le méthotrexate peut être à l’origine de lésions hépatiques chroniques (cirrhose du foie), d’une formation de tissu cicatriciel dans le foie (fibrose hépatique), d’une dégénérescence graisseuse du foie [tous sont des effets peu fréquents], d’une inflammation du foie (hépatite aiguë) [rare] et d’une insuffisance hépatique [très rare] ;
· des symptômes allergiques, comme une éruption cutanée avec démangeaisons et rougeur de la peau, un œdème (gonflement) des mains, des pieds, des chevilles, du visage, des lèvres, de la bouche et de la gorge (pouvant rendre difficile la déglutition ou la respiration) et la sensation de perdre connaissance, ces signes pouvant indiquer une réaction allergique sévère, voire un choc anaphylactique [rare] ;
· des symptômes d’atteinte rénale, comme un œdème (gonflement) des mains, des chevilles ou des pieds ou une modification de la fréquence des mictions ou une diminution (oligurie), voire l’absence (anurie) de production urinaire, ces signes pouvant indiquer une insuffisance rénale [rare] ;
· des symptômes d’une infection, comme une fièvre, des frissons, des douleurs, une inflammation de la gorge, car le méthotrexate peut vous rendre plus sensible aux infections. Des infections sévères, comme un type de pneumonie due à un germe spécifique (pneumonie à Pneumocystis jirovecii) ou un empoisonnement du sang (septicémie) peuvent survenir [rare] ;
· des symptômes tels qu’une faiblesse dans un côté du corps (AVC) ou une douleur, un gonflement, une rougeur et une sensation de chaleur inhabituelle dans l’une de vos jambes (thrombose veineuse profonde) ; cela peut se produire lorsqu’un caillot de sang s’est détaché et bloque un vaisseau sanguin (évènement thromboembolique) [rare] ;
· de la fièvre associée à une aggravation marquée de votre état général ou une fièvre soudaine accompagnée d’une inflammation de la gorge ou de la bouche ou des problèmes urinaires, car le méthotrexate peut être à l’origine d’une chute soudaine du nombre de certains globules blancs (agranulocytose) et d’une dépression sévère de la moelle osseuse [très rare] ;
· des saignements inexpliqués, p. ex. des saignements des gencives, du sang dans les urines, des vomissements de sang ou l’apparition de bleus (ecchymoses), ces signes pouvant indiquer une réduction sévère du nombre de vos plaquettes sanguines due à des épisodes sévères de dépression de la moelle osseuse [très rare] ;
· des symptômes tels que des maux de tête sévères associés à de la fièvre, à une raideur de la nuque, à des nausées, à des vomissements, à une désorientation et à une sensibilité à la lumière peuvent indiquer une inflammation des membranes du cerveau (méningite aseptique aiguë) [très rare] ;
· certains troubles cérébraux (encéphalopathie/leucoencéphalite) ont été signalés chez des patients atteints de cancer recevant un traitement par le méthotrexate. La survenue de tels effets indésirables ne peut pas être exclue lorsque le méthotrexate est utilisé pour traiter d’autres maladies. Les signes de ce type d’affections cérébrales peuvent être une dégradation de l’état mental, des troubles du mouvement (ataxie), des troubles visuels ou des troubles de la mémoire [fréquence indéterminée] ;
· une éruption cutanée très grave ou l’apparition d’ampoules ou de vésicules sur la peau (pouvant également apparaître dans la bouche, au niveau des yeux ou sur les organes génitaux), ces signes pouvant être le signe de syndromes appelés « syndrome de Stevens-Johnson » et « syndrome de brûlure de la peau » (nécrolyse épidermique toxique/syndrome de Lyell) [très rare].
La liste suivante énumère les effets secondaires également susceptibles de se produire :
Très fréquent : peut concerner plus d’1 patient sur 10
· inflammation des muqueuses de la bouche, indigestion, nausées, perte d’appétit, douleurs abdominales ;
· anomalies des tests de la fonction hépatique (ASAT, ALAT, bilirubine, phosphatase alcaline).
Fréquent : peut concerner jusqu’à 1 patient sur 10
· ulcères dans la bouche, diarrhées ;
· éruption cutanée, rougeurs cutanées, démangeaisons ;
· maux de tête, fatigue, somnolence ;
· diminution de la formation de cellules sanguines avec baisse du taux de globules blancs et/ou rouges et/ou de plaquettes.
Peu fréquent : peut concerner jusqu’à 1 patient sur 100
· inflammation de la gorge ;
· inflammation des intestins, vomissements, inflammation du pancréas, selles de couleur noire ou à l’aspect « goudronneux », saignements et ulcères gastro-intestinaux ;
· réactions de type coup de soleil dues à une plus grande sensibilité de la peau au soleil, perte de cheveux, augmentation du nombre de nodosités rhumatismales, ulcère cutané, zona, inflammation des vaisseaux sanguins, éruption cutanée de type herpès, urticaire ;
· apparition d’un diabète ;
· étourdissements, confusion, dépression ;
· baisse du taux d’albumine sérique ;
· diminution du nombre de toutes les cellules sanguines et des plaquettes ;
· inflammation et ulcération de la vessie ou du vagin, diminution de la fonction rénale, troubles de la miction ;
· douleurs articulaires, douleurs musculaires, diminution de la masse osseuse.
Rare : peut concerner jusqu’à 1 patient sur 1 000
· inflammation du tissu gingival ;
· accentuation de la pigmentation de la peau, acné, taches bleues sur la peau (ecchymoses, pétéchies) dues au saignement d’un vaisseau, inflammation des vaisseaux sanguins d’origine allergique ;
· diminution du nombre d’anticorps dans le sang ;
· infection (y compris réactivation d’une infection chronique inactive), yeux rouges (conjonctivite) ;
· sautes d’humeur (altérations de l’humeur) ;
· troubles visuels, pouvant être dus à une lésion des nerfs optiques (neuropathie optique) ;
· inflammation du sac entourant le cœur, accumulation de liquide dans le sac autour du cœur, altération du remplissage cardiaque en raison de la présence de liquide dans le sac autour du cœur ;
· hypotension ;
· formation de tissu cicatriciel dans les poumons (fibrose pulmonaire), essoufflement et asthme bronchique, accumulation de liquide dans le sac entourant les poumons ;
· fracture de fatigue ;
· déséquilibres électrolytiques ;
· fièvre, difficultés de cicatrisation.
Très rare : peut concerner jusqu’à 1 patient sur 10 000
· dilatation toxique aiguë du gros intestin (mégacôlon toxique) ;
· accentuation de la pigmentation des ongles, inflammation des cuticules (panaris aigu), infection profonde des follicules pileux (furonculose), gonflement visible des petits vaisseaux sanguins ;
· douleurs, manque de force ou sensation d’engourdissement ou de fourmillement/sensibilité à la stimulation inférieure à la normale, modifications du goût (goût métallique), convulsions, paralysie, méningisme ;
· baisse de la vision, affection oculaire non inflammatoire (rétinopathie) ;
· diminution du désir sexuel, impuissance, hypertrophie mammaire chez les hommes, production de sperme défectueuse (oligospermie), troubles menstruels, pertes vaginales ;
· hypertrophie des ganglions lymphatiques (lymphome) ;
· syndrome lymphoprolifératif (augmentation excessive des globules blancs).
Fréquence indéterminée : la fréquence ne peut être estimée sur la base des données disponibles
· augmentation du nombre de certains globules blancs ;
· saignements de nez ;
· présence de protéines dans les urines ;
· sensation de faiblesse ;
· lésion osseuse de la mâchoire (secondaire à une augmentation excessive des globules blancs) ;
· destruction de tissus au point d’injection ;
· rougeurs et desquamation de la peau ;
· gonflement.
L’administration sous-cutanée du méthotrexate est bien tolérée localement. Seules des réactions cutanées locales bénignes (comme des sensations de brûlure, un érythème, un œdème, une décoloration, des démangeaisons sévères ou des douleurs), diminuant pendant le traitement, ont été observées.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et le stylo prérempli après EXP. La date de péremption fait référence au dernier jour de ce mois.
À conserver à une température ne dépassant pas 25 °C. Ne pas congeler.
Conserver les stylos préremplis dans l’emballage extérieur, à l’abri de la lumière.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient METOJECT 17,5 mg / 0,35 ml, solution injectable en stylo prérempli
· La substance active est :
Méthotrexate .......... .. 17,5 mg
Pour un stylo prérempli avec 0,35 ml de solution.
1 ml de solution injectable contient 50 mg de méthotrexate.
· Les autres composants sont :
Chlorure de sodium, hydroxyde de sodium et acide chlorhydrique pour ajustement du pH, eau pour préparations injectables.
Ce médicament se présente sous la forme d’une solution injectable en stylo prérempli.
La solution est limpide ; de couleur brun-jaune.
Le stylo prérempli METOJECT est un auto‑injecteur en trois étapes, muni d’un capuchon jaune et d’un bouton poussoir jaune.
Le stylo prérempli METOJECT est un auto‑injecteur en deux étapes, muni d’un capuchon de protection translucide et d’un protège‑aiguille bleu.
METOJECT est disponible en boîtes de 1, 2, 4, 5, 6, 10, 11, 12, 14, 15 et 24 stylos préremplis.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
MEDAC GESELLSCHAFT FÜR KLINISCHE SPEZIALPRÄPARATE MBH
THEATERSTR. 6
22880 WEDEL
ALLEMAGNE
Exploitant de l’autorisation de mise sur le marché
MEDAC S.A.S.
1 RUE CROIX BARRET
69007 LYON
MEDAC GESELLSCHAFT FÜR KLINISCHE SPEZIALPRÄPARATE MBH
THEATERSTR. 6
22880 WEDEL
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Mode d’emploi
Recommandations
Ø Veuillez lire attentivement les instructions ci-dessous avant de procéder à votre injection.
Ø Suivez toujours la technique d’injection expliquée par votre médecin, pharmacien ou infirmier/ère.
Autres sources d’informations
Les procédures pour la manipulation et l’élimination doivent respecter les exigences et réglementations locales. Les femmes enceintes appartenant au personnel soignant ne doivent pas manipuler et/ou administrer METOJECT.
Le méthotrexate ne doit pas entrer en contact avec la peau ou les muqueuses. En cas de contamination, la région affectée doit être rincée immédiatement et abondamment à l’eau.
|
Composants du stylo prérempli METOJECT :
|
|
|
|
a) Avec le capuchon avant l’injection
b) Après avoir retiré le capuchon avant l’injection
c) Après l’injection |
|
Ce que vous devez faire avant de procéder à votre injection 1. Lavez-vous les mains très soigneusement. 2. Sortez le dispositif de son emballage. 3. Vérifiez l’intégrité du stylo prérempli METOJECT avant de l’utiliser.
|
|
|
|
Si le stylo prérempli METOJECT semble avoir été endommagé, ne l’utilisez sous aucun prétexte. Utilisez un autre stylo METOJECT et informez votre médecin, pharmacien ou infirmier/ère de ce problème.
Si une petite bulle d’air est visible dans la zone de contrôle transparente, cela n’a aucun impact sur votre dose et ne constitue aucun danger.
Si vous n’êtes pas en mesure de bien voir ou de vérifier le dispositif de manière suffisamment minutieuse avant l’injection, demandez à l’un de vos proches de vous y aider.
|
|
4. Posez le stylo prérempli METOJECT sur une surface plane et propre (ex. une table). |
|
|
Quelles sont les parties du corps aptes à recevoir l’injection |
|
|
|
Les zones les plus appropriées pour votre injection sont : - le haut des cuisses, - le ventre, à distance du nombril.
· Si l’un de vos proches vous fait l’injection, il est également possible de pratiquer l’injection sur l’arrière de votre bras, juste en dessous de l’épaule. · Alternez les zones d’injection avec chaque injection, ainsi vous réduirez au maximum les risques de réaction au point d’injection. · Ne pratiquez jamais l’injection sur une zone douloureuse, couverte de bleus, de rougeurs ou dure au toucher ou sur une zone présentant des cicatrices ou des vergetures. Si vous souffrez de psoriasis, il est important de ne pas injecter le produit sur une plaque de psoriasis, une zone où la peau est épaissie, rouge ou squameuse ou si elle présente des lésions. |
|
Comment préparer l’injection
|
5. Désinfectez la zone d’injection choisie à l’aide d’un coton imbibé d’alcool.
· Ne retirez pas le capuchon avant d’être prêt(e) à procéder à l’injection. |
|
|
6. Tenez le stylo d’une seule main au niveau de la zone de prise prévue à cet effet, le capuchon doit être dirigé vers le haut. Utilisez l’autre main pour retirer le capuchon doucement dans l’axe du stylo (Ne pas dévisser). L’embout protecteur de l’aiguille se met en place automatiquement dès que le capuchon est retiré. Si cela ne se produit pas, utilisez un autre stylo et informez votre médecin, pharmacien ou infirmier/ère de ce problème. · Si vous n’arrivez pas à retirer le capuchon, demandez à l’un de vos proches de vous y aider. |
|
Remarque : une fois le capuchon retiré, vous devez procéder à l’injection dans les plus brefs délais. |
|
|
|
7. Avec votre main libre, formez un pli cutané entre le pouce et l’index sur la zone choisie pour l’injection une fois nettoyée. · Vous devez maintenir le pli pendant toute la durée de l’injection et ne relâcher la peau qu’une fois l’injection terminée et le stylo retiré. |
|
|
8. Positionnez l’extrémité transparente du stylo prérempli METOJECT (partie où se trouvait initialement le capuchon) perpendiculairement au pli cutané. 9. N’appuyez pas sur le bouton poussoir, mais plaquez le stylo prérempli METOJECT fermement contre votre peau afin de déverrouiller le bouton poussoir. · Si vous n’arrivez pas à positionner le stylo prérempli METOJECT de manière à déverrouiller le bouton poussoir, demandez à l’un de vos proches de vous y aider.
|
|
Comment procéder à l’injection :
|
|
|
|
10. Tout en maintenant fermement le stylo prérempli METOJECT contre la peau, appuyez à présent sur le bouton poussoir avec votre pouce.
11. Vous entendrez un petit déclic indiquant le début de l’injection. Maintenez le stylo en place, sur le pli cutané, jusqu’à ce que l’intégralité du produit ait été injectée. Ce processus peut prendre jusqu’à 5 secondes. |
|
Remarque : Ne retirez pas le stylo prérempli METOJECT de votre peau avant la fin de l’injection, afin d’être sûr(e) que l’intégralité de la dose a été injectée. Si aucun produit n’est injecté, relâchez le bouton et vérifiez que le stylo prérempli METOJECT est bien plaqué contre la peau et appuyez fortement sur le bouton poussoir. Si vous avez des troubles de l’audition, comptez 5 secondes à partir du moment où vous avez actionné le bouton poussoir avant de retirer le stylo prérempli METOJECT du point d’injection. |
|
|
|
12. Retirez le stylo prérempli METOJECT du point d’injection, toujours perpendiculairement à la surface de votre peau (remontez le stylo de manière verticale).
13. L’embout de protection va automatiquement se mettre en place autour de l’aiguille, se verrouiller et vous protéger ainsi de l’aiguille.
14. En cas de léger saignement, mettez un pansement adhésif.
Avant de jeter le stylo prérempli METOJECT, vérifiez qu’il ne reste plus de liquide visible dans le stylo, au fond de la zone de contrôle transparente. Si du liquide reste dans le stylo, c’est que la dose n’a pas totalement été injectée ; consultez alors votre médecin.
|
|
Remarque Afin de vous protéger de toute éventuelle blessure, ne mettez jamais vos doigts sur l’extrémité du stylo contenant l’aiguille. Ne détruisez pas le stylo.
|
|
|
Qui contacter en cas de besoin Ø Pour tout problème ou question, veuillez contacter votre médecin, pharmacien ou infirmier/ère. Ø Si l’un de vos proches est blessé par l’aiguille, consultez votre médecin immédiatement et jetez le stylo prérempli METOJECT. |
|
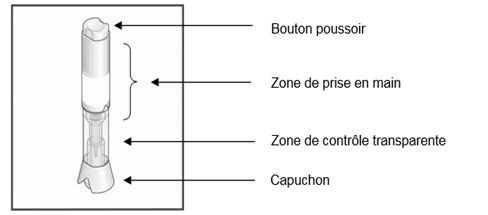
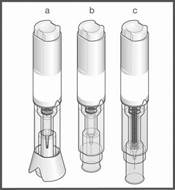
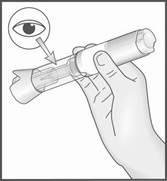
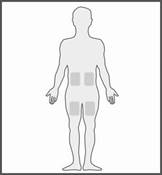
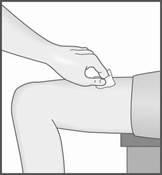
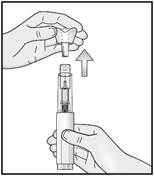
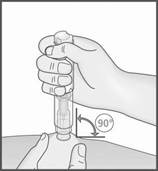
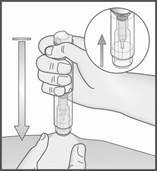
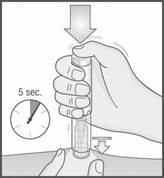
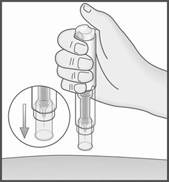
MODE D’EMPLOI
Ce mode d’emploi contient des informations sur la manière de procéder à une injection à l’aide de votre stylo prérempli METOJECT.
Veuillez lire ce mode d’emploi jusqu’à la fin avant d’utiliser le stylo prérempli pour votre injection sous‑cutanée. Veuillez lire le mode d’emploi à chaque renouvellement de votre ordonnance et gardez‑le pour consulter les informations dont vous pourriez avoir besoin après l’utilisation.
Lorsque vous recevez un nouveau médicament ou une nouvelle dose de médicament, vérifiez toujours qu’il/elle correspond à ce que votre médecin vous a prescrit. Avant toute utilisation du stylo, votre professionnel de santé doit vous avoir expliqué, à vous ou à votre aidant, comment vous en servir correctement.
N’utilisez pas le stylo si votre professionnel de santé ne vous a pas appris à vous en servir. Si vous ou votre aidant avez des questions, contactez votre professionnel de santé.
|
Informations importantes à connaître avant de procéder à l’injection avec votre stylo prérempli METOJECT |
|
L’injection à l’aide de votre stylo prérempli METOJECT doit être effectuée une fois par semaine seulement, et toujours le même jour de la semaine.
Vous ne devez pas injecter ou manipuler le produit si vous êtes enceinte.
Conservez le stylo dans un endroit sûr, hors de la vue et de la portée des enfants. En cas de contact avec le médicament, lavez immédiatement et abondamment la zone touchée avec de l’eau.
Ne retirez pas le capuchon de protection avant d’être prêt à procéder à l’injection. Ne partagez pas votre stylo avec une autre personne. N’utilisez pas le stylo si : il est tombé sur une surface dure ou semble endommagé. la solution brun-jaune a changé de couleur, est trouble ou contient des particules. il a été congelé ou conservé à une température supérieure à 25 °C. la date de péremption est dépassée. En cas de doute, contactez votre professionnel de santé. |
Comment conserver votre stylo prérempli METOJECT
A conserver à une température ne dépassant pas 25 °C.
Transportez et conservez le stylo dans l’emballage extérieur, à l’abri de la lumière.
Conservez le stylo dans un endroit sûr, hors de la vue et de la portée des enfants.
Ne pas congeler.
A conserver à une température ne dépassant pas 25 °C.
Le stylo prérempli METOJECT (figure A)
Le stylo prérempli METOJECT est un auto‑injecteur jetable en deux étapes, à dose fixe et à usage unique. Il est disponible en 10 doses différentes allant de 7,5 mg à 30 mg.
|
|
Avant utilisation |
Après utilisation |
|
||
|
Capuchon de protection (translucide) Fenêtre d’inspection (médicament brun-jaune à l’intérieur) Piston (la position diffère selon la dose prescrite)
7,5 mg 15 mg 30 mg (exemples de doses) Zone de code couleur (différent pour chaque dose) |
|
|
(se verrouille après l’injection, aiguille à l’intérieur) Capuchon de (protège‑aiguille à l’intérieur) Tige bleue du piston (indique que la dose a été complètement injectée) Étiquette du produit (pour vérifier la dose et la date de péremption) |


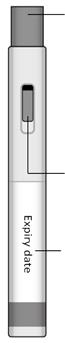
Figure A
Matériel nécessaire à l’injection (figure B)
Le jour de l’injection hebdomadaire, trouvez un endroit confortable, assurez‑vous que la zone est bien éclairée et que vous disposez d’une surface plane et propre, telle qu’une table, pour placer votre matériel d’injection.
Vous avez besoin des éléments suivants :
votre stylo prérempli METOJECT.
Assurez‑vous de disposer également du matériel suivant pour réaliser votre injection (non fourni dans l’emballage) :
votre calendrier pour vérifier le jour de votre injection hebdomadaire ;
un désinfectant pour la peau, tel que de l’alcool, ou à défaut, de l’eau et du savon ;
du coton ou une compresse de gaze pour nettoyer le site d’injection ;
un récipient pour l’élimination du matériel, conformément aux exigences locales.
|
|
||||
|
Calendrier indiquant le jour de l’injection hebdomadaire |
Stylo prérempli METOJECT |
Désinfectant pour la peau |
Coton ou gaze |
Récipient pour l’élimination |
Figure B
|
Préparez‑vous pour votre injection |
|
|||||
|
1. Lavez‑vous les mains et déballez votre stylo (figure C) Lavez‑vous les mains à l’eau et au savon. Sortez le stylo de son emballage avec précaution.
Ne retirez pas le capuchon de protection avant d’être prêt à procéder à l’injection.
|
Figure C
|
|||||
|
2. Vérifiez votre stylo avant de l’utiliser (figure D) Vérifiez soigneusement le nom et la dose sur le stylo et assurez‑vous que vous avez le bon médicament. Si vous ne voyez pas assez bien, demandez de l’aide à quelqu’un.
Vérifiez la date de péremption sur l’étiquette du stylo. N’utilisez pas le stylo s’il est périmé. Vérifiez le médicament à travers la fenêtre d’inspection en retournant le stylo ou en l’agitant doucement. Le médicament à l’intérieur du stylo doit être limpide et de couleur brun-jaune. § N’injectez pas la solution si elle est trouble, a changé de couleur ou si elle contient des particules. § Il est normal de voir une ou plusieurs bulles d’air. N’essayez pas de les éliminer. § Il est possible que vous voyiez une graduation dans la fenêtre ; il n’est pas nécessaire de vous en préoccuper. Vérifiez que le stylo n’est pas endommagé et que le capuchon est bien en place. N’utilisez pas le stylo s’il semble endommagé ou si le capuchon a été retiré ou n’est pas bien fixé.
Si le stylo est périmé, s’il semble endommagé ou s’il n’a pas l’aspect attendu, ne l’utilisez pas et contactez votre professionnel de santé.
Placez délicatement le stylo sur une surface plane et propre, telle qu’une table, avant de passer aux étapes suivantes. |
Figure D
|
|||||
|
3. Choisissez votre site d’injection (figure E) Vous pouvez réaliser l’injection dans : § le haut des cuisses ; § le bas du ventre, en évitant la zone de 5 cm autour du nombril. Si un aidant vous administre l’injection, il peut également utiliser la zone située à l’arrière du haut du bras. Utilisez un site différent de celui de votre dernière injection.
Lorsque vous choisissez un site d’injection : Ne réalisez pas l’injection dans d’autres parties du corps. Ne réalisez pas l’injection dans une région de peau qui présente un bleu, qui est sensible, qui pèle ou qui est rouge ou dure. Ne réalisez pas l’injection dans des grains de beauté, des cicatrices ou des vergetures. Ne réalisez pas l’injection au travers des vêtements. Site réservé à l’aidant Site réservé à l’aidant Figure E
|
|
|||||
|
4. Nettoyez le site d’injection (figure F) Nettoyez le site d’injection avec un désinfectant à l’alcool ; à défaut, vous pouvez utiliser de l’eau et du savon. Laissez sécher la peau à l’air libre.
Ne soufflez pas sur la zone nettoyée. Ne touchez plus le site d’injection tant que vous n’avez pas terminé votre injection. |
Figure F |
|||||
|
Injection de la dose |
|
|||||
|
5. Retirez le capuchon (figure G) Retirez le capuchon de protection uniquement lorsque vous êtes prêt à procéder à l’injection.
N’essayez pas de remettre le capuchon sur le stylo une fois qu’il a été retiré.
Tenez le stylo avec le capuchon dirigé vers le haut et tirez fermement sur le capuchon pour le retirer. Tirez tout droit, sans incliner ni faire tourner le capuchon, pour le retirer. Jetez le capuchon immédiatement. Il est possible que de petites gouttelettes de médicament apparaissent. Ceci est normal. Effectuez votre injection immédiatement après avoir retiré le capuchon.
Ne touchez pas le protège‑aiguille bleu avec les doigts, car cela pourrait déclencher accidentellement l’injection et vous blesser.
|
Figure G |
|||||
|
6. Positionnez votre stylo (figure H) Placez le protège‑aiguille bleu décapuchonné sur la peau à la verticale (90 degrés), la fenêtre d’inspection tournée vers vous pour que vous puissiez la voir. Vous pourriez vous sentir plus à l’aise en pinçant délicatement la peau autour du site d’injection entre votre pouce et votre index avant l’injection, mais cela n’est pas nécessaire avec ce stylo.
|
Figure H |
|||||
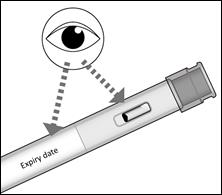

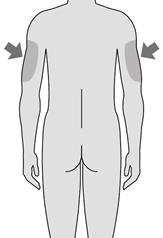
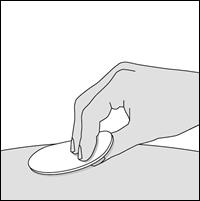
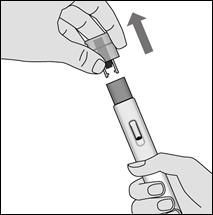
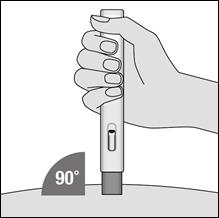
|
7. Commencez l’injection (figure I) Pour commencer l’injection, pressez le stylo à fond contre la peau. Cela fera glisser le protège‑aiguille bleu vers le haut à l’intérieur du stylo et l’injection commencera automatiquement. Un premier déclic indique le début de l’injection. La tige bleue du piston descendra alors. Maintenez le stylo contre la peau jusqu’à ce que la totalité du médicament ait été injectée. Ne changez pas la position du stylo une fois que l’injection a commencé.
|
1er déclic Figure I |
||||
|
8. Maintenez le stylo en place jusqu’à la fin de l’injection (Figure J) Continuez à presser le stylo contre votre peau. L’injection est terminée lorsque : ü vous entendez un deuxième déclic, peu après le premier ; ü ou : la tige bleue du piston a cessé de se déplacer et remplit la fenêtre d’inspection ; ü ou : 5 secondes se sont écoulées.
Ne retirez pas le stylo avant qu’au moins 5 secondes se soient écoulées. |
2e déclic 5 sec. Figure J
|
||||
|
9. Fin de l’injection (figure K) Retirez le stylo du site d’injection en le relevant à la verticale. Le protège‑aiguille bleu recouvre automatiquement l’aiguille. Le protège‑aiguille bleu est ensuite verrouillé. Vérifiez qu’il ne reste pas de médicament brun-jaune à l’intérieur de la fenêtre d’inspection. Si vous voyez encore du médicament brun-jaune à l’intérieur de la fenêtre, il se peut que vous n’ayez pas reçu toute la dose. Si cela se produit ou si vous avez d’autres inquiétudes, contactez votre professionnel de santé.
Ne touchez pas le protège‑aiguille bleu après l’injection. Vous pourriez vous blesser. |
Figure K
|


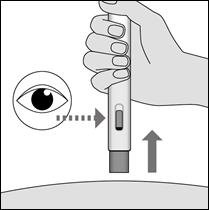
|
Après votre injection |
||
|
10. Nettoyez le site d’injection (figure L) Vous remarquerez peut‑être une petite goutte de sang au niveau du site d’injection. Ceci est normal. Si nécessaire, appuyez sur la zone avec du coton ou une compresse de gaze. Au besoin, recouvrez le site d’injection à l’aide d’un petit pansement.
Ne frottez pas le site d’injection.
|
Figure L |
|
|
11. Jetez votre stylo (figure M) Chaque stylo ne peut être utilisé qu’une seule fois. Ne remettez pas le capuchon en place sur le stylo.
Placez le stylo usagé et son capuchon de protection hors de la vue et de la portée des enfants.
Jetez le capuchon et le stylo immédiatement après utilisation. Le médicament et le stylo prérempli doivent être éliminés conformément aux exigences locales. Jetez le reste du matériel usagé à la poubelle, avec les ordures ménagères. Le carton peut être recyclé.
Éliminez de façon sécurisée les stylos préremplis METOJECT qui sont périmés, qui ne sont plus nécessaires ou qui sont inutilisables pour d’autres raisons. |
Figure M |
|