Dernière mise à jour le 01/06/2026
OLANZAPINE VIATRIS 5 mg, comprimé orodispersible
Indications thérapeutiques
Classe pharmacothérapeutique : diazépines, oxazépines et thiazépines - code ATC N05AH03.
OLANZAPINE VIATRIS contient le principe actif olanzapine qui appartient à une famille de médicaments appelés antipsychotiques.
OLANZAPINE VIATRIS aussi est utilisé pour traiter des épisodes maniaques modérés et sévères, des troubles de l’humeur pouvant être caractérisés, entre autres, par un sentiment d’euphorie, une activité et énergie excessive, une diminution du besoin de sommeil, le fait de parler trop vite avec une accélération des idées et parfois une irritabilité sévère.
Il est également un régulateur de l’humeur qui prévient la survenue des états invalidants d’euphorie ou de dépression associés à cette pathologie.
Présentations
> 28 plaquette(s) polyamide aluminium PVC de 1 comprimé(s)
Code CIP : 276 038-2 ou 34009 276 038 2 2
Déclaration de commercialisation : 17/09/2015
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 17,42 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 18,44 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 27/03/2025
OLANZAPINE VIATRIS 5 mg, comprimé orodispersible
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé orodispersible contient 5 mg d'olanzapine.
Excipients à effet notoire :
Chaque comprimé orodispersible contient 1,975 mg d'aspartame.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé jaune à jaune clair, uni à moucheté, rond, plat, à bords biseautés, gravé « M » sur une face et « OE1 » sur l'autre face.
4.1. Indications thérapeutiques
L’olanzapine est indiquée dans le traitement de la schizophrénie.
Chez les patients ayant initialement répondu au traitement, l’olanzapine a démontré son efficacité à maintenir cette amélioration clinique au long cours.
L'olanzapine est indiquée dans le traitement des épisodes maniaques modérés à sévères.
L’olanzapine est indiquée dans la prévention des récidives chez les patients présentant un trouble bipolaire, ayant déjà répondu au traitement par l’olanzapine lors d’un épisode maniaque (voir rubrique 5.1).
4.2. Posologie et mode d'administration
Adultes
Schizophrénie : la dose initiale recommandée d’olanzapine est de 10 mg par jour.
Épisode maniaque : la dose initiale est de 15 mg par jour en une seule prise en monothérapie ou 10 mg par jour en association (voir rubrique 5.1).
Prévention des récidives chez les patients présentant un trouble bipolaire : la dose initiale recommandée est de 10 mg/jour. Chez les patients traités par l’olanzapine lors d’un épisode maniaque, pour la prévention des récidives, le traitement sera maintenu à la même dose. Si un nouvel épisode (maniaque, mixte ou dépressif) survient, le traitement par olanzapine doit être poursuivi (à la posologie optimale). Selon l’expression clinique de l’épisode, un traitement de la symptomatologie thymique sera associé.
Dans le traitement de la schizophrénie, épisodes maniaques, prévention des récidives chez les patients présentant un trouble bipolaire, la posologie journalière peut être adaptée en fonction de l’état clinique du patient entre 5 et 20 mg par jour. Une augmentation à des doses plus importantes que la dose initiale recommandée n'est conseillée qu'après une réévaluation clinique appropriée et ne doit généralement être envisagée qu'à intervalles de 24 heures minimum.
Sujets âgés
Une dose initiale plus faible (5 mg par jour) n’est pas indiquée de façon systématique mais doit être envisagée chez les patients âgés de 65 ans et plus lorsque des facteurs cliniques le justifient (voir rubrique 4.4).
Insuffisance rénale et/ou hépatique
Une dose initiale plus faible (5 mg) doit être envisagée pour ces patients. En cas d’insuffisance hépatique modérée (cirrhose, Child-Pugh de classe A ou B), la dose initiale devra être de 5 mg et sera augmentée uniquement avec précaution.
Fumeurs
La dose initiale et l’intervalle de doses ne nécessitent pas d’adaptation chez les non-fumeurs par rapport aux fumeurs. Le métabolisme de l’olanzapine peut être stimulé par le tabagisme. Une surveillance clinique est recommandée et une augmentation de la posologie de l’olanzapine peut être envisagée, si nécessaire (voir rubrique 4.5).
L’existence de plus d’un facteur pouvant ralentir le métabolisme (sexe féminin, sujet âgé, non-fumeur) peut justifier une réduction de la dose initiale. Lorsqu’elle est indiquée, l’augmentation posologique sera faite avec précaution chez ces patients.
Si une progression posologique de 2,5 mg est nécessaire, les comprimés pelliculés d’olanzapine doivent être utilisés (Voir rubriques 4.5 et 5.2).
Population pédiatrique
L’utilisation de l’olanzapine chez les enfants et les adolescents âgés de moins de 18 ans n’est pas recommandée du fait du manque de données sur la sécurité d’emploi et l’efficacité. Une prise de poids, des anomalies lipidiques et des taux de prolactine ont été rapportés selon une ampleur plus élevée dans les études à court terme chez les patients adolescents comparativement aux études chez les patients adultes (voir rubriques 4.4, 4.8, 5.1 et 5.2).
Mode d’administration
L'olanzapine peut être administrée pendant ou en dehors des repas, la prise de nourriture n'ayant pas d'incidence sur l'absorption. Il convient de diminuer progressivement les doses lors de l’arrêt de l’olanzapine.
OLANZAPINE VIATRIS, comprimé orodispersible se casse facilement, vous devez ainsi le manipuler avec précaution. Ne pas manipuler les comprimés avec des mains humides car les comprimés pourraient se casser.
Pour les plaquettes prédécoupées, tenir la plaquette par un bord et séparer une cellule du reste de la plaquette en la détachant délicatement suivant la ligne de prédécoupage. Détacher avec soin du support.
Pour les plaquettes non prédécoupées, retirer délicatement le film aluminium en faisant attention de ne pas retirer le film aluminium des alvéoles adjacentes. Pousser doucement le comprimé.
OLANZAPINE VIATRIS, comprimé orodispersible doit être placé dans la bouche où il sera rapidement dissous dans la salive et donc facilement avalé. Une fois dans la bouche, il est difficile de retirer intact le comprimé orodispersible. Le comprimé orodispersible étant friable, il doit être administré immédiatement après ouverture de la plaquette. Il peut être également dissous dans un grand verre d'eau ou dans toute autre boisson adaptée (jus d'orange, jus de pomme, lait ou café) immédiatement avant administration.
Il a été démontré que le comprimé orodispersible d'olanzapine est bio-équivalent aux comprimés pelliculés d’olanzapine, avec un taux et un niveau d'absorption similaires. La posologie et la fréquence d'administration de cette forme sont identiques à celles des comprimés pelliculés d’olanzapine. Le comprimé orodispersible d’olanzapine peut être utilisé comme une alternative à la forme comprimés pelliculés d’olanzapine.
· Hypersensibilité à la substance active ou à l’un des excipients, mentionnés à la rubrique 6.1.
· Patients présentant un risque connu de glaucome à angle fermé.
4.4. Mises en garde spéciales et précautions d'emploi
Démence accompagnée de troubles psychotiques et/ou troubles du comportement
L’olanzapine n’est pas recommandée pour une utilisation chez les patients présentant une démence accompagnée de troubles psychotiques et/ou des troubles du comportement du fait d’une augmentation du risque de mortalité et d’accidents vasculaires cérébraux. Au cours d’essais cliniques contrôlés versus placebo (durée de 6 à 12 semaines), réalisés chez des patients âgés (âge moyen 78 ans) souffrant de démence accompagnée de troubles psychotiques et/ou de troubles du comportement, l’incidence des décès dans le groupe olanzapine a été deux fois plus importante que celle observée dans le groupe placebo (3,5 versus 1,5 % respectivement). L’incidence plus élevée de décès n’a pas été corrélée à la dose d’olanzapine (dose moyenne quotidienne de 4,4 mg) ou à la durée de traitement. Dans cette population de patients, un âge supérieur à 65 ans, une dysphagie, une sédation, une malnutrition et une déshydratation, une pathologie pulmonaire (telle qu’une pneumopathie avec ou sans inhalation) ou une utilisation concomitante de benzodiazépines peuvent être des facteurs prédisposant à une augmentation du risque de mortalité.
Néanmoins, indépendamment de ces facteurs de risque, l’incidence de mortalité a été supérieure dans le groupe olanzapine (comparativement au placebo).
Des événements indésirables vasculaires cérébraux (tels qu’accidents vasculaires cérébraux, accidents ischémiques transitoires), dont certains à issue fatale, ont été rapportés dans ces mêmes essais cliniques. Trois fois plus d’événements indésirables vasculaires cérébraux ont été rapportés dans le groupe de patients traités par olanzapine comparativement au groupe de patients traités par placebo (1,3 % versus 0,4 % respectivement). Tous les patients traités par olanzapine ou par placebo ayant présenté un événement vasculaire cérébral avaient des facteurs de risque préexistants. Un âge supérieur à 75 ans et une démence de type vasculaire ou mixte ont été identifiés comme des facteurs de risque d’événements indésirables vasculaires cérébraux dans le groupe olanzapine. L’efficacité de l’olanzapine n’a pas été démontrée dans ces essais.
Maladie de Parkinson
L’utilisation de l’olanzapine pour traiter la psychose associée aux agonistes dopaminergiques chez les patients parkinsoniens n’est pas recommandée. Au cours d’essais cliniques, une aggravation de la symptomatologie parkinsonienne et des hallucinations ont été très fréquemment rapportées et de façon plus fréquente qu’avec le placebo (voir rubrique 4.8) et l’olanzapine n’était pas plus efficace que le placebo dans le traitement des symptômes psychotiques. Dans ces essais, les patients devaient être stabilisés en début d’étude avec la posologie minimale efficace du traitement antiparkinsonien (agoniste dopaminergique) et poursuivre le même traitement antiparkinsonien, au même dosage, pendant toute l’étude. La posologie initiale de l’olanzapine était de 2,5 mg/jour, puis pouvait être ajustée par l’investigateur jusqu’à un maximum de 15 mg/jour.
Syndrome Malin des Neuroleptiques (SMN)
Le Syndrome Malin des Neuroleptiques (SMN) est un syndrome potentiellement mortel associé aux traitements antipsychotiques. De rares cas rapportés comme un Syndrome Malin des Neuroleptiques (SMN) ont également été notifiés sous olanzapine. Les signes cliniques du SMN sont l'hyperthermie, la rigidité musculaire, l’altération des facultés mentales, et des signes d'instabilité neuro-végétative (instabilité du pouls et de la pression artérielle, tachycardie, hypersudation et troubles du rythme cardiaque). Peuvent s'ajouter des signes tels qu’élévation de la créatine phosphokinase, myoglobinurie (rhabdomyolyse) et insuffisance rénale aiguë. Si un patient présente des signes ou des symptômes évoquant un SMN, ou une hyperthermie inexpliquée non accompagnée d’autres manifestations cliniques de SMN, tous les médicaments antipsychotiques, y compris l’olanzapine, doivent être arrêtés.
Hyperglycémie et diabète
Des cas d’hyperglycémie et/ou de survenue ou exacerbation d’un diabète, associés parfois à une acidocétose ou un coma, avec une issue fatale pour certains cas, ont été rapportés de manière peu fréquente (voir rubrique 4.8). Dans certains cas, une prise de poids antérieure, qui pourrait être un facteur prédisposant, a été rapportée. Une surveillance clinique appropriée est souhaitable conformément aux recommandations en vigueur sur les antipsychotiques, par exemple mesurer la glycémie au début du traitement par olanzapine, 12 semaines après l’instauration du traitement, puis tous les ans.
Les patients traités par des médicaments antipsychotiques, incluant l’olanzapine, doivent être surveillés afin de détecter les signes et symptômes d’une hyperglycémie (tels que polydipsie, polyurie, polyphagie et faiblesse) et les patients ayant un diabète de type II ou des facteurs de risque de diabète de type II doivent être suivis régulièrement pour surveiller la détérioration du contrôle de la glycémie.
Le poids doit être surveillé régulièrement, par exemple au début du traitement, 4, 8 et 12 semaines après l’instauration du traitement par olanzapine, puis tous les 3 mois.
Anomalies lipidiques
Des anomalies lipidiques ont été observées chez des patients traités par l’olanzapine au cours d’essais cliniques versus placebo (voir rubrique 4.8). Les modifications lipidiques doivent être prises en charge de façon appropriée sur le plan clinique, notamment chez les patients présentant des troubles lipidiques et chez les patients ayant des facteurs de risque pouvant favoriser le développement de troubles lipidiques. Le bilan lipidique des patients traités par des médicaments antipsychotiques, incluant l’olanzapine, doit être surveillé régulièrement conformément aux recommandations en vigueur sur les antipsychotiques, par exemple au début du traitement, 12 semaines après l’instauration du traitement par olanzapine, puis tous les 5 ans.
Activité anticholinergique
Bien que l’olanzapine ait montré une activité anticholinergique in vitro, l’incidence des effets liés à cette activité a été faible au cours des essais cliniques. Cependant, l’expérience clinique de l’olanzapine étant limitée chez les patients ayant une pathologie associée, la prudence est recommandée lors de sa prescription chez des patients présentant une hypertrophie prostatique, un iléus paralytique ou toute autre pathologie en rapport avec le système cholinergique.
Fonction hépatique
Des élévations transitoires et asymptomatiques des aminotransférases (ALAT et ASAT) ont été fréquemment observées, notamment en début de traitement. La prudence s'impose chez les patients présentant une élévation des ALAT et/ou des ASAT, chez les patients présentant des signes et des symptômes évocateurs d'une atteinte hépatique, chez les patients atteints d’affections préexistantes associées à une réserve fonctionnelle limitée et chez les patients traités par des médicaments potentiellement hépatotoxiques et un suivi doit être instauré. Dans les cas où une hépatite a été diagnostiquée (comprenant des atteintes hépatiques cytolytiques, cholestatiques ou mixtes), le traitement par olanzapine doit être arrêté.
Neutropénie
La prudence s'impose chez les patients dont le nombre de leucocytes et/ou de neutrophiles est faible quelle qu'en soit la cause, chez les patients recevant des médicaments connus pour induire des neutropénies, chez les patients ayant des antécédents de dépression médullaire ou de myélotoxicité médicamenteuse, chez les patients atteints de dépression médullaire causée par une pathologie intercurrente, une radiothérapie ou une chimiothérapie, et chez les patients atteints d'hyperéosinophilie ou de syndromes myéloprolifératifs. Des neutropénies ont été fréquemment rapportées lors de l’administration concomitante de l'olanzapine et du valproate (voir rubrique 4.8).
Arrêt du traitement
Des symptômes aigus tels que sueurs, insomnie, tremblements, anxiété, nausées ou vomissements ont été rarement rapportés (≥ 0,01 % et < 0,1 %) lors de l’arrêt brutal du traitement par olanzapine.
Intervalle QT
Au cours des essais cliniques, un allongement du QTc cliniquement significatif (QT corrigé selon la formule de Fridericia [QTcF] ≥ 500 millisecondes [msec] à n’importe quel moment après l’inclusion chez les patients ayant à l’inclusion un QTcF < 500 msec) a été rapporté de manière peu fréquente (0,1 % à 1 %) chez les patients traités par olanzapine, sans différence significative par rapport au placebo quant aux événements cardiaques associés. Cependant, la prudence est recommandée lors de la co-prescription avec des médicaments connus pour allonger l’intervalle QTc, notamment chez le sujet âgé ou chez des patients présentant un syndrome de QT long congénital, une insuffisance cardiaque congestive, une hypertrophie cardiaque, une hypokaliémie ou une hypomagnésémie.
Atteintes thrombo-emboliques
Des atteintes thrombo-emboliques veineuses ont été rapportées de manière peu fréquente avec l’olanzapine (≥ 0,1 %, < 1 %). Il n’a pas été établi de lien de causalité entre la survenue de ces atteintes et le traitement par olanzapine.
Cependant, les patients schizophrènes présentant souvent des facteurs de risque thrombo-embolique veineux, tout facteur de risque potentiel d’atteintes thrombo-emboliques veineuses (telle immobilisation des patients) doit être identifié et des mesures préventives mises en œuvre.
Activité générale sur le Système Nerveux Central
Compte tenu des principaux effets de l’olanzapine sur le Système Nerveux Central, il faudra être prudent lors de l’association avec des médicaments à action centrale et avec l’alcool. Du fait de son activité antagoniste de la dopamine in vitro, l’olanzapine peut antagoniser les effets des agonistes directs et indirects de la dopamine.
Convulsions
L’olanzapine doit être utilisée avec prudence chez les patients qui ont des antécédents de convulsions ou qui sont placés dans des conditions susceptibles d’abaisser leur seuil convulsif. Des cas de convulsions ont été rapportés de manière peu fréquente chez les patients traités par olanzapine. Dans la plupart de ces cas, il existait soit des antécédents de convulsions soit des facteurs de risque de convulsions.
Dyskinésie tardive
Dans les études comparatives de durée inférieure ou égale à un an, l’incidence des dyskinésies liées au traitement a été significativement plus faible dans le groupe olanzapine. Cependant, le risque de survenue de dyskinésie tardive augmentant avec la durée de l’exposition, la réduction posologique, voire l’arrêt du traitement, doit être envisagée dès l’apparition de signes ou symptômes de dyskinésie tardive chez les patients sous olanzapine. Ces symptômes peuvent provisoirement s’aggraver ou même survenir après l’arrêt du traitement.
Hypotension orthostatique
Une hypotension orthostatique a été rarement observée chez les sujets âgés lors des essais cliniques.
Il est recommandé de mesurer périodiquement la pression artérielle des patients de plus de 65 ans.
Mort subite d’origine cardiaque
Depuis la commercialisation de l’olanzapine, des cas de mort subite d’origine cardiaque ont été rapportés chez les patients traités avec l’olanzapine. Dans une étude de cohorte observationnelle rétrospective, le risque de mort subite présumée d’origine cardiaque chez les patients traités avec l’olanzapine a été environ le double du risque existant chez les patients ne prenant pas d’antipsychotiques. Dans cette étude, le risque avec l’olanzapine a été comparable au risque avec des antipsychotiques atypiques inclus dans une analyse groupée.
Population pédiatrique
L’olanzapine n’est pas indiquée chez les enfants et les adolescents. Des études réalisées chez des patients âgés de 13 à 17 ans ont montré divers événements indésirables, incluant prise de poids, modification des paramètres métaboliques et élévations des taux sanguins de prolactine (voir rubriques 4.8 et 5.1).
Excipients
OLANZAPINE VIATRIS, comprimé orodispersible contient de l'aspartame, source de phénylalanine. L’aspartame est hydrolysé dans le tube gastro‑digestif lorsqu’il est ingéré par voie orale. L’un des principaux produits de l’hydrolyse est la phénylalanine. Peut être dangereux pour les personnes atteintes de phénylcétonurie (PCU), une maladie génétique rare caractérisée par l’accumulation de phénylalanine ne pouvant être éliminée correctement.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé orodispersible, c.‑à‑d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les études d’interaction ont été réalisées uniquement chez l’adulte.
Interactions potentielles ayant un effet sur l'olanzapine
L'olanzapine étant métabolisée par le cytochrome CYP1A2, les produits qui stimulent ou inhibent spécifiquement cette iso-enzyme peuvent modifier les paramètres pharmacocinétiques de l'olanzapine.
Induction du CYP1A2
Le métabolisme de l’olanzapine peut être stimulé par le tabagisme et la carbamazépine, ce qui peut entraîner une diminution des concentrations de l'olanzapine. Seule une augmentation légère à modérée de la clairance de l'olanzapine a été observée. Il est probable que les conséquences cliniques soient limitées, mais une surveillance clinique est recommandée et une augmentation de la posologie de l'olanzapine peut être envisagée, si nécessaire (voir rubrique 4.2).
Inhibition du CYP1A2
Il a été montré que la fluvoxamine, inhibiteur spécifique du CYP1A2, inhibe significativement le métabolisme de l’olanzapine. La fluvoxamine entraîne une augmentation moyenne de la Cmax de l’olanzapine de 54 % chez les femmes non fumeuses et de 77 % chez les hommes fumeurs.
L’augmentation moyenne de l’ASC de l’olanzapine était respectivement de 52 % et de 108 %. Une posologie initiale plus faible de l’olanzapine doit être envisagée chez les patients traités par la fluvoxamine ou tout autre inhibiteur du CYP1A2 comme la ciprofloxacine. Une diminution de la posologie de l’olanzapine doit être envisagée si un traitement par un inhibiteur du CYP1A2 est instauré.
Diminution de la biodisponibilité
Le charbon activé diminue la biodisponibilité de l’olanzapine par voie orale de 50 à 60 % et doit être pris au moins 2 heures avant ou après l'administration de l'olanzapine.
Avec la fluoxétine (inhibiteur du CYP2D6), des doses uniques d'anti-acides (aluminium, magnésium) ou la cimétidine, il n'a pas été retrouvé d'effet significatif sur les paramètres pharmacocinétiques de l'olanzapine.
Effets potentiels de l’olanzapine sur les autres médicaments
L'olanzapine peut antagoniser les effets directs et indirects des agonistes dopaminergiques.
L'olanzapine n'inhibe pas les principales iso-enzymes du CYP450 in vitro (c'est-à-dire 1A2, 2D6, 2C9, 2C19, 3A4). Par conséquent, aucune interaction particulière n'est attendue comme cela a pu être vérifié lors d'études in vivo au cours desquelles aucune inhibition du métabolisme des produits actifs suivants n'a été mise en évidence : antidépresseurs tricycliques (représentant principalement la voie du CYP2D6), la warfarine (CYP2C9), la théophylline (CYP1A2) ou le diazépam (CYP3A4 et 2C19).
Aucune interaction n'a été mise en évidence lors de la prise concomitante de l'olanzapine et du lithium ou du bipéridène.
Le suivi des taux plasmatiques du valproate n’a pas montré la nécessité d’adapter la posologie du valproate après l’instauration d’un traitement par l’olanzapine.
Activité générale sur le Système Nerveux Central
La prudence est recommandée chez les patients qui consomment de l’alcool ou qui sont traités par des médicaments dépresseurs du système nerveux central.
L’utilisation concomitante de l’olanzapine et de médicaments antiparkinsoniens chez les patients atteints de la maladie de Parkinson et de démence est déconseillée (voir rubrique 4.4).
Intervalle QTc
La prudence s’impose si l’olanzapine est administrée de manière concomitante avec des médicaments connus pour allonger l’intervalle QTc (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
Aucune étude contrôlée spécifique n’a été réalisée chez la femme enceinte. Les patientes doivent être averties de la nécessité d’informer leur médecin de toute grossesse ou tout désir de grossesse au cours du traitement par l’olanzapine. Cependant, l’expérience chez la femme étant limitée, l’olanzapine ne doit être administrée pendant la grossesse que si les bénéfices potentiels justifient les risques fœtaux potentiels.
Les nouveau-nés exposés aux antipsychotiques (dont l’olanzapine) pendant le troisième trimestre de la grossesse présentent un risque de réactions indésirables incluant des symptômes extrapyramidaux et/ou des symptômes de sevrage pouvant varier en termes de sévérité et de durée après l’accouchement.
Les réactions suivantes ont été rapportées : agitation, hypertonie, hypotonie, tremblements, somnolence, détresse respiratoire, trouble de l’alimentation. En conséquence, les nouveau-nés doivent être étroitement surveillés.
Dans une étude chez des femmes volontaires qui allaitaient, l’olanzapine a été retrouvée dans le lait maternel. L’exposition moyenne des nouveau-nés à l’état d’équilibre (en mg/kg) a été estimée à environ 1,8 % de la dose d’olanzapine reçue par la mère (en mg/kg). L’allaitement maternel est donc déconseillé aux patientes en cours de traitement par olanzapine.
Fertilité
Les effets sur la fertilité ne sont pas connus (voir les informations précliniques mentionnées à la rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de tolérance
Adultes
Les effets indésirables les plus fréquemment rapportés (≥ 1 % des patients) associés à l’utilisation d’olanzapine au cours des essais cliniques ont été : somnolence, prise de poids, éosinophilie, augmentation des taux de prolactine, de cholestérol, de la glycémie et de la triglycéridémie (voir rubrique 4.4), glucosurie, augmentation de l'appétit, sensation vertigineuse, akathisie, parkinsonisme, leucopénie, neutropénie (voir rubrique 4.4), dyskinésie, hypotension orthostatique, effets anticholinergiques, élévations transitoires asymptomatiques des aminotransférases hépatiques (voir rubrique 4.4), rash, asthénie, fatigue, fièvre, arthralgie, phosphatase alcaline augmentée, gamma-glutamyltransférase augmentée, uricémie augmentée, créatine phosphokinase augmentée et œdème.
La liste des effets indésirables présentés dans le tableau suivant a été établie à partir du recueil des événements indésirables et des examens de laboratoire issus de la notification spontanée et des essais cliniques.
Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Les catégories de fréquence sont définies ainsi : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Fréquence indéterminée |
|
|
Affections hématologiques et du système lymphatique |
|||||
|
Éosinophilie Leucopénie10 Neutropénie10 |
Thrombocytopénie11 |
|
|||
|
Affections du système immunitaire |
|||||
|
Hypersensibilité11 |
|
||||
|
Troubles du métabolisme et de la nutrition |
|||||
|
Prise de poids1 |
Augmentation de la cholestérolémie2, 3 Augmentation de la glycémie4 Augmentation de la triglycéridémie 2, 5 Glucosurie Augmentation de l'appétit |
Survenue ou exacerbation d’un diabète, associée parfois à une acidocétose ou un coma, avec une issue fatale pour certains cas (voir rubrique 4.4)11 |
Hypothermie12 |
|
|
|
Affections du système nerveux |
|||||
|
Somnolence |
Vertiges Akathisie6 Parkinsonisme6 Dyskinésie6 |
Convulsions avec, dans la plupart des cas, des antécédents de convulsions ou bien des facteurs de risque de convulsions rapportés11. Dystonie (incluant des crises oculogyres) 11 Dyskinésie tardive11 Amnésie9 Dysarthrie Bégaiement11 Syndrome des Jambes Sans Repos11 |
Syndrome Malin des Neuroleptiques (voir rubrique 4.4)12 Symptômes à l’arrêt du traitement 7,12 |
|
|
|
Affections cardiaques |
|||||
|
Bradycardie Allongement du QTc (voir rubrique 4.4) |
Tachycardie/fibrillation ventriculaire Mort subite (voir rubrique 4.4)11 |
|
|||
|
Affections vasculaires |
|||||
|
Hypotension orthostatique10 |
Atteinte thrombo-embolique (comprenant embolie pulmonaire et thrombose veineuse profonde) (voir rubrique 4.4) |
|
|||
|
Affections respiratoires, thoraciques et médiastinales |
|||||
|
Épistaxis9 |
|
||||
|
Affections gastro-intestinales |
|
||||
|
Effets anticholinergiques légers et transitoires tels que constipation et bouche sèche |
Distension abdominale9 Hypersécrétion salivaire11 |
Pancréatite11 |
|
||
|
Affections hépatobiliaires |
|||||
|
Élévations transitoires et asymptomatiques des aminotransférases hépatiques (ASAT, ALAT), particulièrement en début de traitement (voir rubrique 4.4). |
Hépatite (comprenant des atteintes hépatiques hépatocellulaire, cholestatiques ou mixtes)11 |
|
|||
|
Affections de la peau et du tissu sous-cutané |
|||||
|
Rash |
Réaction de photosensibilité Alopécie |
Syndrome d'hypersensibilité médicamenteuse (DRESS) |
|||
|
Affections musculo-squelettiques et systémiques |
|||||
|
Arthralgie9 |
Rhabdomyolyse11 |
|
|||
|
Affections du rein et des voies urinaires |
|||||
|
|
|
Incontinence urinaire Rétention urinaire Retard à la miction11 |
|
|
|
|
Affections gravidiques, puerpérales et périnatales |
|||||
|
|
Syndrome de sevrage médicamenteux néonatal (voir rubrique 4.6) |
||||
|
Affections des organes de reproduction et du sein |
|||||
|
Dysfonction érectile chez les hommes Diminution de la libido chez les hommes et les femmes |
Aménorrhée Gonflement mammaire Galactorrhée chez les femmes Gynécomastie/gonflement mammaire chez les hommes |
Priapisme12 |
|
||
|
Troubles généraux et anomalies au site d'administration |
|||||
|
Asthénie Fatigue Œdème Fièvre10 |
|
||||
|
Investigations |
|||||
|
Augmentation de la prolactinémie8 |
Phosphatase alcaline augmentée10 Créatine phosphokinase augmentée11 Gamma-glutamyl-transférase augmentée10 Uricémie augmentée10 |
Augmentation de la bilirubine totale |
|
||
1 Une prise de poids cliniquement significative a été observée dans toutes les catégories d’Indice de Masse Corporelle (IMC) de départ. Après un traitement de courte durée (durée médiane de 47 jours), une augmentation de poids supérieure ou égale à 7 % par rapport au poids initial a été très fréquente (22,2 %), une augmentation de poids supérieure ou égale à 15 % par rapport au poids initial a été fréquente (4,2 %) et une augmentation de poids supérieure ou égale à 25 % par rapport au poids initial a été peu fréquente (0,8 %). Une augmentation de poids supérieure ou égale à 7 %, à 15 % et à 25 % par rapport au poids initial a été très fréquente (64,4 %, 31,7 % et 12,3 % respectivement) lors d’une utilisation prolongée (au moins 48 semaines).
2 Les augmentations moyennes des taux lipidiques à jeun (cholestérol total, cholestérol LDL et triglycérides) ont été plus élevées chez les patients sans signes de trouble des lipides au début du traitement.
3 Observée pour des taux à jeun normaux au début du traitement (< 5,17 mmol/l) qui sont devenus élevés (≥ 6,2 mmol/l). Une augmentation des taux de cholestérol total à jeun ayant une valeur limite au début du traitement (≥ 5,17- < 6,2 mmol/l) à des valeurs élevées (≥ 6,2 mmol/l) a été très fréquente.
4 Observée pour des taux à jeun normaux au début du traitement (< 5,56 mmol/l) qui sont devenus élevés (≥ 7 mmol/l). Une augmentation des taux de glucose à jeun ayant une valeur limite au début du traitement (≥ 5,56 - < 7 mmol/l) à des valeurs élevées (≥ 7 mmol/l) a été très fréquente.
5 Observée pour des taux à jeun normaux au début du traitement (< 1,69 mmol/l) qui sont devenus élevés (≥ 2,26 mmol/l). Une augmentation des taux de triglycérides à jeun ayant une valeur limite au début du traitement (≥ 1,69 - < 2,26 mmol/l) à des valeurs élevées (≥ 2,26 mmol/l) a été très fréquente.
6 Au cours d'essais cliniques, l'incidence des troubles parkinsoniens et des dystonies dans le groupe olanzapine était numériquement supérieure à celle du groupe placebo (pas de différence statistique significative). Les patients traités par l'olanzapine ont présenté une plus faible incidence de troubles parkinsoniens, d’akathisie et de dystonie que les patients traités par l’halopéridol à des posologies comparables. En l’absence d’information précise concernant les antécédents de mouvements anormaux extrapyramidaux de survenue aiguë ou tardive, on ne peut conclure à ce jour que l’olanzapine entraîne moins de dyskinésies tardives et/ou de syndromes extrapyramidaux tardifs.
7 Des symptômes aigus tels que sueurs, insomnie, tremblements, anxiété, nausées et vomissements ont été rapportés lors de l’arrêt brutal du traitement par olanzapine.
8 Dans des études cliniques allant jusqu’à 12 semaines, une prolactinémie dépassant la limite supérieure de la normale a été observée chez environ 30 % des patients traités avec l’olanzapine et ayant un taux de prolactine normal au début du traitement. Chez la majorité de ces patients, les augmentations étaient généralement légères et sont restées inférieures à deux fois la limite supérieure de la normale.
9 Effet indésirable identifié à partir de la base de données des essais cliniques intégrant l’olanzapine.
10 Telles qu’évaluées grâce aux valeurs mesurées à partir de la base de données des essais cliniques intégrant l’olanzapine.
11 Effet indésirable identifié à partir de la notification spontanée dont la fréquence est déterminée en utilisant la base de données intégrant l’olanzapine.
12 Effet indésirable identifié à partir de la notification spontanée dont la fréquence est estimée à la limite de l’intervalle de confiance à 95 % en utilisant la base de données intégrant l’olanzapine.
Utilisation prolongée (au moins 48 semaines)
La proportion de patients ayant présenté des modifications indésirables cliniquement significatives du poids (augmentation), du glucose, du cholestérol total/HDL/LDL ou des triglycérides a augmenté au cours du temps. Chez les patients adultes qui ont suivi 9-12 mois de traitement, le taux d’augmentation de la glycémie sanguine moyenne a diminué après 6 mois environ.
Information complémentaire concernant des populations particulières
Au cours d’essais cliniques chez des patients âgés déments, le traitement par olanzapine a été associé à une incidence supérieure de décès et d’événements indésirables vasculaires cérébraux par rapport au placebo (voir rubrique 4.4). Une démarche anormale et des chutes ont été des événements indésirables très fréquemment rapportés avec l’olanzapine. Des pneumonies, une augmentation de la température corporelle, une léthargie, un érythème, des hallucinations visuelles et des incontinences urinaires ont été fréquemment observés.
Au cours d’essais cliniques menés chez des patients parkinsoniens souffrant de psychoses médicamenteuses (agonistes dopaminergiques), une aggravation de la symptomatologie parkinsonienne et des hallucinations ont été très fréquemment rapportées et ce, de façon plus fréquente, qu’avec le placebo.
Au cours d'un essai clinique mené chez des patients atteints de manie bipolaire, lors de la prise concomitante de valproate et d’olanzapine la fréquence des neutropénies a été de 4,1 % ; un facteur contributif potentiel pourrait être des taux plasmatiques élevés de valproate. Une augmentation supérieure > à 10 % des cas de tremblements, bouche sèche, augmentation de l'appétit et prise de poids a été observée lors de l'association de l'olanzapine au lithium ou au valproate. Des troubles de l'élocution ont également été fréquemment rapportés. Lors de l'association de l'olanzapine au lithium ou au divalproex une augmentation supérieure ou égale à 7 % du poids initial est survenue chez 17,4 % des patients pendant la phase aiguë du traitement (jusqu'à 6 semaines). Lors du traitement au long cours par l’olanzapine (jusqu’à 12 mois) dans la prévention des récidives chez les patients présentant un trouble bipolaire, une augmentation de poids supérieure ou égale à 7 % par rapport au poids initial a été rapportée chez 39,9 % des patients.
Population pédiatrique
L’olanzapine n’est pas indiquée chez les enfants et adolescents âgés de moins de 18 ans. Bien qu’aucune étude clinique comparant les adolescents aux adultes n’ait été réalisée, les données issues des études réalisées chez l’adolescent ont été comparées à celles issues des essais chez l’adulte.
Le tableau suivant résume les effets indésirables rapportés avec une fréquence plus importante chez les patients adolescents (âgés de 13 à 17 ans) que chez les patients adultes ou les effets indésirables uniquement observés lors des essais cliniques de courte durée réalisés chez les patients adolescents.
Une prise de poids cliniquement significative (≥ 7 %) surviendrait plus fréquemment chez les adolescents comparés à des patients adultes avec une exposition comparable. L’amplitude de la prise de poids et la proportion des patients adolescents qui ont eu une augmentation du poids cliniquement significative ont été plus importantes lors d’une exposition prolongée (au moins 24 semaines) que lors d’une exposition de courte durée.
Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Les catégories de fréquence sont définies ainsi : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10).
Troubles du métabolisme et de la nutrition
Très fréquent : prise de poids13, augmentation de la triglycéridémie14, augmentation de l'appétit.
Fréquent : augmentation de la cholestérolémie15.
Affections du système nerveux
Très fréquent : sédation (dont hypersomnie, léthargie, somnolence).
Affections gastro-intestinales
Fréquent : bouche sèche.
Affections hépatobiliaires
Très fréquent : élévations des aminotransférases hépatiques (ASAT, ALAT ; voir rubrique 4.4).
Investigations
Très fréquent : diminution de la bilirubine totale, augmentation de la Gamma Globuline Transférase, augmentation de la prolactinémie16.
13 Après un traitement de courte durée (durée médiane de 22 jours), une augmentation de poids supérieure ou égale à 7 % par rapport au poids initial (kg) a été très fréquente (40,6 %), une augmentation de poids supérieure ou égale à 15 % par rapport au poids initial a été fréquente (7,1 %) et une augmentation de poids supérieure ou égale à 25 % par rapport au poids initial a été fréquente (2,5 %). Lors d’une exposition prolongée (au moins 24 semaines), 89,4 % des patients ont eu une augmentation du poids supérieure ou égale à 7 %, 55,3 % ont eu une augmentation de poids supérieure ou égale à 15 % et 29,1 % ont eu une augmentation de poids supérieure ou égale à 25 % par rapport à leur poids initial.
14 Observée pour des taux à jeun normaux au début du traitement (< 1,016 mmol/l) qui sont devenus élevés (≥ 1,467 mmol/l) et des modifications des taux de triglycérides à jeun ayant une valeur limite au début du traitement (≥ 1,016 - < 1,467 mmol/l) devenant élevée (≥ 1,467 mmol/l).
15 Des modifications des taux de cholestérol total à jeun ayant une valeur normale au début du traitement (< 4,39 mmol/l) devenant élevée (≥ 5,17 mmol/l) a été fréquente. Des modifications des taux de cholestérol total à jeun ayant une valeur limite au début du traitement (≥ 4,39 - < 5,17 mmol/l) devenant élevée (≥ 5,17 mmol/l) ont été très fréquentes.
16 Augmentation de la prolactinémie rapportée chez 47,4 % des patients adolescents.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Signes et symptômes
En cas de surdosage, les symptômes très fréquemment observés (incidence > 10 %) sont : tachycardie, agitation/agressivité, dysarthrie, symptômes extrapyramidaux divers et diminution du niveau de conscience allant de la sédation au coma.
Les autres effets cliniquement significatifs du surdosage sont : délire, convulsions, coma, éventuel syndrome malin des neuroleptiques, insuffisance respiratoire, fausse route, hypertension ou hypotension, arythmies cardiaques (moins de 2 % des cas de surdosage) et arrêt cardio-respiratoire.
Des évolutions fatales ont été rapportées pour des surdosages aigus à une dose aussi basse que 450 mg mais une évolution favorable a également été rapportée à la suite d’un surdosage aigu par environ 2 g d’olanzapine orale.
Conduite à tenir
Il n’y a pas d’antidote spécifique de l’olanzapine. Il n’est pas recommandé de provoquer des vomissements. La prise en charge standard d'un surdosage peut être utilisée (à savoir lavage gastrique, administration de charbon activé). L’administration concomitante de charbon activé réduit la biodisponibilité orale de l’olanzapine de 50 à 60 %.
Un traitement symptomatique et une surveillance des fonctions vitales doivent être mis en œuvre selon l’état clinique, y compris un traitement de l’hypotension et du collapsus circulatoire et une assistance respiratoire. Ne pas utiliser l’adrénaline, la dopamine ou un autre bêta-sympathomimétique car la stimulation des récepteurs bêta-adrénergiques peut aggraver l’hypotension. Un monitoring cardiovasculaire est nécessaire pour déceler d’éventuelles arythmies. Une surveillance médicale étroite et le monitoring doivent être poursuivis jusqu’à la guérison du patient.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Effets pharmacodynamiques
L’olanzapine est un agent antipsychotique, un traitement antimaniaque et thymorégulateur avec un large profil pharmacologique sur un certain nombre de récepteurs.
Dans les études précliniques, l’olanzapine a montré une affinité pour certains récepteurs (Ki < 100 nM) tels que les récepteurs sérotoninergiques 5-HT2A/2C, 5-HT3, 5-HT6, dopaminergiques D1, D2, D3, D4, D5, muscariniques cholinergiques M1-M5, α1 adrénergiques et histaminiques H1. Des études de comportement chez l’animal avec l’olanzapine ont montré un antagonisme des systèmes 5-HT, dopaminergiques et cholinergiques, ce qui confirme le profil de liaison aux récepteurs. Il a été démontré dans des études in vitro que l’olanzapine avait une plus grande affinité pour les récepteurs sérotoninergiques 5-HT2 que pour les récepteurs dopaminergiques D2, et une plus grande activité in vivo sur les modèles 5-HT2 par rapport aux modèles D2. Il a été démontré par des études électrophysiologiques que l’olanzapine réduit de façon sélective la transmission au niveau des neurones dopaminergiques du système mésolimbique (A10) alors que l’effet observé sur le système striatal (A9) impliqué dans l’activité motrice est limité. L’olanzapine réduit la réponse d’évitement conditionné, test qui peut indiquer une activité antipsychotique, à des doses inférieures à celles responsables d’induction de catalepsie, effet qui peut indiquer la survenue d’effets indésirables moteurs. Contrairement à d’autres agents antipsychotiques, l’olanzapine augmente la réponse à un test « d’anxiolyse ».
Dans une étude de tomographie par émission de positron (PET) chez le volontaire sain utilisant une dose orale unique (10 mg), l’olanzapine a entraîné une occupation des récepteurs 5-HT2A supérieure à celle des récepteurs D2. De plus, une étude d’imagerie en tomographie par émission monophotonique (SPECT) chez des patients schizophrènes a mis en évidence une occupation du système striatal D2 plus faible chez les patients répondant à l’olanzapine que chez les patients répondant à d’autres antipsychotiques et à la rispéridone, et comparable à celle observée chez des patients répondant à la clozapine.
Efficacité et sécurité clinique
Dans les deux études versus placebo et dans deux études sur trois réalisées versus produits de référence chez 2 900 patients schizophrènes présentant à la fois une symptomatologie positive et négative, l’olanzapine a été associée à une amélioration de la symptomatologie positive et négative statistiquement plus importante.
Dans un essai international comparatif en double aveugle ayant inclus 1 481 patients présentant des troubles schizophréniques ou schizoaffectifs ou apparentés, associés à des symptômes dépressifs d’intensités variables (score initial à l’échelle de dépression de Montgomery et Asberg de 16,6), une analyse prospective dont un critère secondaire de jugement était l’évolution de la symptomatologie dépressive avant - après traitement a mis en évidence une amélioration statistiquement plus importante (p = 0,001) dans le groupe de traitement olanzapine (- 6,0) que dans le groupe de traitement halopéridol (-3,1).
Chez les patients présentant un épisode maniaque ou mixte dans le cadre de troubles bipolaires, l'olanzapine a montré une efficacité supérieure à celle du placebo et du valproate monosodique (dilvaproex) sur la réduction des symptômes maniaques sur 3 semaines. L'olanzapine a également montré des résultats d'efficacité comparables à l'halopéridol en termes de proportion de patients en rémission des symptômes maniaques et dépressifs à 6 et 12 semaines. Dans une étude chez des patients traités par le lithium ou le valproate depuis au moins deux semaines, l'introduction de 10 mg d'olanzapine (en association avec le lithium ou le valproate) a entraîné, après 6 semaines, une réduction des symptômes maniaques supérieure à celle observée chez les patients traités par le lithium ou le valproate en monothérapie.
Lors d’une étude de prévention des récurrences de 12 mois, réalisée chez des patients présentant un épisode maniaque, ayant obtenu une rémission suite au traitement par olanzapine et ayant subi une randomisation vers le groupe olanzapine ou le groupe placebo, l’olanzapine présentait une supériorité statistiquement significative par rapport au placebo, concernant le critère d’évaluation primaire de récurrence bipolaire. L’olanzapine présentait également un avantage statistiquement significatif par rapport au placebo en termes de prévention des récurrences tant de la manie que de la dépression.
Une deuxième étude de12 mois sur la prévention des récidives a été menée chez des patients qui présentaient des épisodes maniaques et qui étaient parvenus au stade de la rémission après un traitement associant l’olanzapine avec le lithium. Ceux-ci ont ensuite été randomisés à l’olanzapine ou au lithium seul. L’olanzapine s’est montrée statistiquement non inférieure au lithium sur le taux de récidive, critère principal de l’étude (olanzapine 30,0 %, lithium 38,3 % ; p=0,055).
Dans une étude comparative à 18 mois chez des patients présentant un épisode maniaque ou mixte stabilisés après un traitement associant l’olanzapine avec un thymorégulateur (lithium ou valproate), le traitement à long terme associant l’olanzapine au lithium ou au valproate ne présentait pas une supériorité statistiquement significative par rapport au lithium ou au valproate seul dans le délai de survenue d’un épisode bipolaire, défini en fonction des critères syndromique (de diagnostic).
Population pédiatrique
Les données comparatives d’efficacité chez les adolescents (âgés de 13 à 17 ans) sont limitées à des études à court terme dans la schizophrénie (6 semaines) et la manie associée à des troubles bipolaires de type I (3 semaines), impliquant moins de 200 adolescents. L’olanzapine a été utilisée à une dose flexible démarrant à 2,5 mg et allant jusqu’à 20 mg par jour. Durant le traitement par l’olanzapine, les adolescents ont pris de manière significative plus de poids comparativement aux adultes. L’ampleur des modifications des taux à jeun de cholestérol total, de cholestérol LDL, de triglycérides et de prolactine (voir rubriques 4.4 et 4.8) était plus importante chez les adolescents que chez les adultes. Il n’y a pas de données sur le traitement de maintien et sur la sécurité à long terme (voir rubriques 4.4 et 4.8). Les informations sur la sécurité d’emploi à long terme sont principalement limitées à des données non contrôlées en ouvert.
5.2. Propriétés pharmacocinétiques
Absorption
L’olanzapine est bien absorbée après administration orale, les concentrations plasmatiques maximales étant atteintes dans un délai de 5 à 8 heures. L’absorption n’est pas influencée par la présence d’aliments. La biodisponibilité orale absolue par rapport à l’administration intraveineuse n’a pas été déterminée.
Distribution
Le taux de fixation de l’olanzapine aux protéines plasmatiques est d’environ 93 %, pour une fourchette de concentration allant d’environ 7 à 1 000 ng/ml. L’olanzapine se lie essentiellement à l’albumine et à l’α1-glycoprotéine acide.
Biotransformation
L’olanzapine est métabolisée dans le foie par conjugaison et oxydation. Le principal métabolite circulant est le 10-N-glucuronide ; il ne franchit pas la barrière hémato-encéphalique. Les cytochromes P450-CYP1A2 et P450-CYP2D6 entraînent la formation du métabolite N-desméthyl et du métabolite 2-hydroxyméthyl. Ces deux métabolites ont montré une activité pharmacologique in vivo significativement plus faible que l’olanzapine dans les études animales.
L'activité pharmacologique principale est due à la molécule mère, l’olanzapine.
Élimination
Après administration orale, la demi-vie moyenne d’élimination terminale de l’olanzapine chez le sujet sain varie selon l’âge et le sexe.
Chez le sujet sain âgé (65 ans et plus), par rapport au sujet sain jeune, la demi-vie moyenne d’élimination de l’olanzapine est prolongée (51,8 versus 33,8 heures) et la clairance est réduite (17,5 vs 18,2 l/heure). La variabilité pharmacocinétique chez le sujet âgé est comparable à celle observée chez le sujet jeune. Chez 44 patients schizophrènes et âgés de plus de 65 ans, des doses de 5 à 20 mg par jour n’ont pas été associées à un profil d’effets indésirables particulier.
Chez la femme, par rapport à l’homme, la demi-vie d’élimination moyenne est légèrement prolongée (36,7 vs 32,3 heures) et la clairance est réduite (18,9 vs 27,3 l/heure). Cependant, l’olanzapine (5-20 mg) a montré un profil de sécurité comparable chez la femme (n=467) et chez l’homme (n=869).
Insuffisant rénal
Chez les patients atteints d’insuffisance rénale (clairance de la créatinine < 10 ml/min), par rapport aux sujets sains, ni la demi-vie d’élimination moyenne (37,7 vs 32,4 heures), ni la clairance (21,2 vs 25,0 l/heure) ne sont significativement différentes. Toutefois, des études du bilan de masse ont montré qu’environ 57 % d’une dose d’olanzapine marquée par un isotope radioactif ont été excrétés dans les urines, principalement sous forme de métabolites.
Insuffisance hépatique
Une petite étude sur l’effet de l’altération de la fonction hépatique chez 6 sujets présentant une cirrhose cliniquement significative (Child-Pugh de classe A (n = 5) et B (n = 1)) a démontré un léger effet sur la pharmacocinétique de l’olanzapine administrée par voie orale (dose unique de 2,5 ‑ 7,5 mg). Les sujets présentant une dysfonction hépatique légère à modérée avaient une clairance systémique légèrement augmentée et une demi-vie d’élimination plus rapide par rapport aux sujets ne présentant pas de dysfonction hépatique (n = 3). Il y avait plus de fumeurs parmi les sujets présentant une cirrhose (4/6 ; 67 %) que parmi les sujets ne présentant pas de dysfonction hépatique (0/3 ; 0 %).
Tabagisme
Chez les non-fumeurs, par rapport aux fumeurs (hommes et femmes), la demi-vie d’élimination est prolongée (38,6 vs 30,4 heures) et la clairance est réduite (18,6 vs 27,7 l/heure).
La clairance plasmatique de l’olanzapine est plus faible chez les sujets âgés que chez les sujets jeunes, chez les femmes que chez les hommes, et chez les non-fumeurs que chez les fumeurs. Toutefois, l’impact de l’âge, du sexe ou du tabagisme sur la clairance et la demi-vie de l’olanzapine est faible par rapport à la variabilité globale interindividuelle.
Une étude comprenant des sujets caucasiens, japonais et chinois n’a montré aucune différence dans les paramètres pharmacocinétiques entre les trois populations.
Population pédiatrique
Adolescents (âgés de 13 à 17 ans) : les paramètres pharmacocinétiques de l'olanzapine sont similaires entre les adolescents et les adultes. Dans des études cliniques, la moyenne d’exposition à l’olanzapine était approximativement supérieure de 27 % chez les adolescents. Les différences démographiques entre les adolescents et les adultes concernent un poids corporel moyen inférieur et un nombre moins important de fumeurs chez les adolescents. De tels facteurs pourraient contribuer à l’observation de la moyenne d’exposition plus élevée chez les adolescents.
5.3. Données de sécurité préclinique
Les signes de toxicité après administration orale chez les rongeurs sont caractéristiques des neuroleptiques puissants : hypo-activité, coma, tremblements, convulsions cloniques, salivation, et diminution de la prise de poids. Les doses médianes létales étaient d’environ 210 mg/kg (souris) et 175 mg/kg (rats). Les chiens ont toléré des doses orales uniques allant jusqu’à 100 mg/kg sans décéder. Les signes cliniques observés ont été les suivants : sédation, ataxie, tremblements et accélération de la fréquence cardiaque, respiration difficile, myosis et anorexie. Chez le singe, des doses orales uniques allant jusqu’à 100 mg/kg ont entraîné une prostration et, à des doses supérieures, un état de semi-inconscience.
Toxicité à doses répétées
Dans des études d’une durée allant jusqu’à 3 mois chez la souris et jusqu’à 1 an chez le rat et le chien, les effets essentiels ont été une dépression du SNC, des effets anticholinergiques et des troubles hématologiques périphériques. Une tolérance est apparue pour la dépression du SNC. Les paramètres de croissance ont été diminués aux fortes doses. Les effets réversibles liés à l’augmentation de la prolactinémie chez la rate comprenaient une diminution du poids des ovaires et de l’utérus, des modifications morphologiques de l’épithélium vaginal et de la glande mammaire.
Toxicité hématologique
Des effets hématologiques ont été observés dans chacune des espèces, y compris des diminutions dose-dépendantes du nombre des leucocytes circulants chez la souris et une diminution non spécifique des leucocytes circulants chez le rat ; cependant, aucun signe de cytotoxicité médullaire n’a été mis en évidence.
Une neutropénie réversible, une thrombocytopénie ou une anémie sont survenues chez quelques chiens traités par 8 ou 10 mg/kg/j (l’exposition totale à l’olanzapine [ASC] étant 12 à 15 fois plus élevée que celle d’un homme ayant reçu une dose de 12 mg). Chez des chiens cytopéniques, aucun effet indésirable sur les cellules souches ou prolifératives de la moelle osseuse n'a été observé.
Toxicité de la reproduction
L’olanzapine n’a montré aucun effet tératogène. La sédation a eu un effet sur la capacité d’accouplement des rats mâles. Les cycles œstraux ont été affectés aux doses de 1,1 mg/kg (soit 3 fois la posologie maximale chez l'Homme) et les paramètres de reproduction ont été influencés chez les rats ayant reçu des doses de 3 mg/kg (9 fois la posologie maximale chez l'Homme). Dans les portées de rats ayant reçu de l’olanzapine, un retard du développement fœtal et une diminution transitoire du taux d’activité des petits ont été observés.
Mutagénicité
L’olanzapine n’a montré aucun effet mutagène ni clastogène, lors d’une série complète de tests standard, tels que tests de mutation bactérienne, et tests in vitro et in vivo sur mammifères.
Carcinogénicité
D’après les résultats des études chez la souris et le rat, il a été conclu que l’olanzapine n’est pas carcinogène.
2 ans.
6.4. Précautions particulières de conservation
À conserver dans l’emballage d'origine à l’abri de la lumière et de l’humidité.
6.5. Nature et contenu de l'emballage extérieur
Flacon (PEHD) muni d'un bouchon à vis en polypropylène avec un joint d'étanchéité, coton absorbant et desséchant (gel de silice). Flacon de 7, 10, 14, 28, 30, 56, 98, 100, 250 ou 500 comprimés.
Plaquette (OPA/AI/PVC). Boîte de 7, 10, 14, 28, 30, 35, 56, 60, 70, 98 ou 100 comprimés.
Plaquette prédécoupée unitaire (OPA/AI/PVC). Boîte de (7x1), (10x1), (14x1), (28x1), (30x1), (35x1), (56x1), (60x1), (70x1), (98x1) ou (100x1) comprimés.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1 RUE DE TURIN
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 276 035 3 2 : 28 comprimés orodispersibles sous plaquettes (OPA/Al/PVC).
· 34009 276 037 6 1 : 30 comprimés orodispersibles sous plaquettes (OPA/Al/PVC).
· 34009 276 038 2 2 : 28x1 comprimés orodispersibles sous plaquettes unitaire (OPA/Al/PVC).
· 34009 276 039 9 0 : 30x1 comprimés orodispersibles sous plaquettes unitaire (OPA/Al/PVC).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Médicament soumis à prescription médicale.
Liste I
ANSM - Mis à jour le : 27/03/2025
OLANZAPINE VIATRIS 5 mg, comprimé orodispersible
Olanzapine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce qu’OLANZAPINE VIATRIS 5 mg, comprimé orodispersible et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre OLANZAPINE VIATRIS 5 mg, comprimé orodispersible ?
3. Comment prendre OLANZAPINE VIATRIS 5 mg, comprimé orodispersible ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver OLANZAPINE VIATRIS 5 mg, comprimé orodispersible ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE OLANZAPINE VIATRIS 5 mg, comprimé orodispersible ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : diazépines, oxazépines et thiazépines - code ATC N05AH03.
OLANZAPINE VIATRIS contient le principe actif olanzapine qui appartient à une famille de médicaments appelés antipsychotiques.
OLANZAPINE VIATRIS aussi est utilisé pour traiter des épisodes maniaques modérés et sévères, des troubles de l’humeur pouvant être caractérisés, entre autres, par un sentiment d’euphorie, une activité et énergie excessive, une diminution du besoin de sommeil, le fait de parler trop vite avec une accélération des idées et parfois une irritabilité sévère.
Il est également un régulateur de l’humeur qui prévient la survenue des états invalidants d’euphorie ou de dépression associés à cette pathologie.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE OLANZAPINE VIATRIS 5 mg, comprimé orodispersible ?
Ne prenez jamais OLANZAPINE VIATRIS 5 mg, comprimé orodispersible
· si vous êtes allergique à l’olanzapine ou à l’un des autres composants contenus dans ce médicament (mentionnés à la rubrique 6). Une réaction allergique peut prendre la forme d’une éruption cutanée, de démangeaisons, de gonflement de la face, des lèvres, de la langue, de la gorge, de difficulté à respirer ou d’essoufflement. Si vous avez déjà éprouvé de telles manifestations, vous devez en informer votre médecin ;
· si on vous a préalablement diagnostiqué des problèmes oculaires tels que certains types de glaucome (augmentation de la pression intra-oculaire).
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant de prendre OLANZAPINE VIATRIS, comprimé orodispersible :
· si vous ou quelqu’un d’autre dans votre famille avez des antécédents de caillots sanguins car les médicaments de cette classe ont été associés à la formation de caillots sanguins ;
· si vous êtes un patient âgé souffrant de démence car elle peut entraîner des effets indésirables graves ;
· si vous avez du diabète ;
· si vous avez une maladie cardiaque ;
· si on vous a informé que vous avez un déséquilibre hydro-électrolytique dans le sang (en particulier, un taux bas de potassium ou de magnésium) ;
· si vous êtes né avec un intervalle QT prolongé (objectivé à l’ECG, un enregistrement électrique du cœur) ;
· si vous avez des problèmes de foie ou de reins ;
· si vous avez la maladie de Parkinson ;
· si vous avez des antécédents de convulsions ou de crises d’épilepsie ;
· si vous avez un élargissement de la prostate ;
· si vous souffrez de constipation importante (iléus paralytique) ;
· si vous avez une quantité faible de globules blancs (qui peut être causée par des médicaments, une radiothérapie, une chimiothérapie ou une maladie de la moelle osseuse) ;
· si on vous a informé que vous avez un nombre élevé de certains globules blancs dans le sang ou une maladie de la moelle osseuse dite « myéloproliférative » entraînant une augmentation excessive du nombre de cellules sanguines ;
· en cas d’accident vasculaire cérébral ou d’accident ischémique transitoire ;
· si vous fumez (la dose d’olanzapine pourra être ajustée).
Pendant le traitement
Si vous présentez une association d’une très forte fièvre, d’une respiration accélérée, une sudation excessive, un changement d’humeur, une rigidité musculaire, une augmentation de la pression sanguine, et une somnolence ou un endormissement, parlez-en à votre médecin qui peut décider d’arrêter OLANZAPINE VIATRIS.
Si vous présentez des mouvements involontaires de la face ou de la langue, parlez-en à votre médecin car il peut envisager de diminuer ou d’arrêter OLANZAPINE VIATRIS.
Une prise de poids a été observée chez des patients prenant de l’olanzapine. Vous et votre médecin devez vérifier votre poids régulièrement. Envisagez de vous orienter vers un diététicien ou une aide avec un régime alimentaire si nécessaire.
Des taux élevés de sucre et de graisses (triglycérides et cholestérol) dans le sang ont été observés chez des patients prenant de l’olanzapine. Votre médecin devra réaliser des tests sanguins afin de vérifier les taux de sucre et de certaines graisses dans votre sang avant que vous ne commenciez à prendre ce médicament.
À titre de précaution, si vous avez plus de 65 ans, votre pression artérielle peut être contrôlée par votre médecin.
Enfants et adolescents
OLANZAPINE VIATRIS n’est pas recommandé chez les patients de moins de 18 ans.
Autres médicaments et OLANZAPINE VIATRIS 5 mg, comprimé orodispersible
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament. En particulier, prévenez votre médecin si vous prenez l’un des médicaments suivants :
· des médicaments utilisés pour traiter la maladie de Parkinson ;
· des antidépresseurs ou des médicaments pour l'anxiété ou l'insomnie (tranquillisants) car ils peuvent entraîner une somnolence ;
· de la carbamazépine (utilisée comme anti-épileptique ou stabilisateur de l’humeur) ;
· de la fluvoxamine (un antidépresseur) ;
· de la ciprofloxacine (un antibiotique) ;
· des médicaments qui peuvent modifier votre rythme cardiaque comme des anti-arythmiques (tels que l’amiodarone, le sotalol, la quinidine, la disopyramide), des antibiotiques (qui appartiennent à la classes des macrolides), des antidépresseurs tricycliques ;
· du charbon activé (une substance chimique utilisée pour absorber d’autres médicaments), il doit être pris 2 heures au moins avant ou après la prise d’olanzapine car il peut interférer avec l’absorption d’olanzapine.
OLANZAPINE VIATRIS 5 mg, comprimé orodispersible avec des aliments, boissons et de l’alcool
Ne buvez pas d'alcool pendant le traitement par OLANZAPINE VIATRIS car l'association de ce médicament avec l'alcool peut entraîner une somnolence.
Si vous êtes enceinte ou que vous si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou à votre pharmacien avant de prendre ce médicament. Ce médicament ne doit pas vous être prescrit si vous allaitez, car de faibles quantités d’olanzapine peuvent passer dans le lait maternel.
Les symptômes suivants peuvent apparaître chez les nouveau-nés dont les mères ont utilisé de l’olanzapine durant le dernier trimestre (les trois derniers mois de leur grossesse) : tremblements, raideur et/ou faiblesse musculaire, endormissement, agitation, problème de respiration et difficulté à s’alimenter. Si votre bébé développe l’un de ces symptômes, vous devez contacter votre médecin.
Conduite de véhicules et utilisation de machines
Le traitement par OLANZAPINE VIATRIS comporte un risque de somnolence ou d’étourdissement. Si cela survient, ne conduisez pas ou n’utilisez pas certains outils ou machines. Informez votre médecin.
Aspartame
OLANZAPINE VIATRIS 5 mg, comprimé orodispersible contient 1,975 mg d’aspartame par comprimé. L’aspartame contient une source de phénylalanine. Peut être dangereux pour les personnes atteintes de phénylcétonurie (PCU), une maladie génétique rare caractérisée par l’accumulation de phénylalanine ne pouvant être éliminée correctement.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé orodispersible, c.‑à‑d. qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE OLANZAPINE VIATRIS 5 mg, comprimé orodispersible ?
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Votre médecin vous indiquera combien de comprimés d’OLANZAPINE VIATRIS vous devez prendre et pendant combien de temps. La dose journalière recommandée d’OLANZAPINE VIATRIS se situe entre 5 et 20 mg.
Consultez votre médecin si vos symptômes réapparaissent mais n’arrêtez pas de prendre votre médicament sauf nouvelle indication de votre médecin.
OLANZAPINE VIATRIS doit être pris une fois par jour, conformément à la prescription de votre médecin. Efforcez-vous de prendre le ou les comprimés à la même heure tous les jours. Vous pouvez les prendre avec ou sans nourriture. Les comprimés orodispersibles d’OLANZAPINE VIATRIS sont utilisés par voie orale.
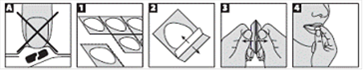
Les comprimés d’OLANZAPINE VIATRIS se cassent facilement. Il convient donc de les manipuler avec soin. Ne prenez pas les comprimés avec des mains humides car les comprimés peuvent s'effriter.
1. Pour les plaquettes prédécoupées pour dose unitaire, tenir la plaquette par un bord et séparer une cellule du reste de la plaquette en la détachant délicatement en suivant la ligne de prédécoupage.
2. Détacher avec soin du support. Pour les plaquettes non prédécoupées, retirer délicatement le film aluminium en faisant attention de ne pas retirer le film aluminium des alvéoles adjacentes.
3. Pousser doucement le comprimé.
4. Mettre le comprimé dans la bouche. Il se dissoudra directement dans votre bouche et pourra alors être aisément avalé.
Au lieu de mettre le comprimé dans votre bouche, vous pouvez également l'ajouter à un grand verre d'eau, de jus d'orange, de jus de pomme, de lait ou de café et remuer. Avec certaines boissons, le mélange peut changer de couleur et éventuellement devenir trouble. Le boire immédiatement.
Si vous avez pris plus d’OLANZAPINE VIATRIS 5 mg, comprimé orodispersible que vous n’auriez dû
Contactez votre médecin ou votre hôpital immédiatement. Montrez-lui votre boîte de comprimés.
Les patients ayant pris plus d’olanzapine qu’ils n’auraient dû ont présenté les signes suivants : accélération du rythme cardiaque, agitation/agressivité, problèmes d’élocution, mouvements anormaux (particulièrement du visage et de la langue), diminution du niveau de conscience. Les autres signes peuvent être : confusion aiguë, convulsions (épilepsie), coma, association de fièvre, d’une accélération de la respiration, de sueurs, de raideur musculaire, de somnolence ou d’une envie de dormir, d’une diminution de la fréquence respiratoire, inhalation de fluide dans la trachée et les poumons (« fausse route »), d’une pression artérielle élevée ou basse, de troubles du rythme cardiaque.
Si vous oubliez de prendre OLANZAPINE VIATRIS 5 mg, comprimé orodispersible
Si vous arrêtez de prendre OLANZAPINE VIATRIS 5 mg, comprimé orodispersible
N’arrêtez pas de prendre vos comprimés simplement parce que vous vous sentez mieux. Il est important que vous preniez OLANZAPINE VIATRIS aussi longtemps que votre médecin vous l’aura indiqué.
Si vous arrêtez brutalement de prendre OLANZAPINE VIATRIS, des symptômes tels que sueurs, difficulté à dormir, tremblements, anxiété ou sensation d’être malade (nausées) et devenir malade (vomissements) peuvent survenir. Votre médecin peut donc vous demander de réduire les doses progressivement avant d’arrêter le traitement.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Si vous remarquez l’un des effets indésirables suivants, allez immédiatement aux urgences de l’hôpital le plus proche ou informez votre médecin immédiatement :
Fréquent (pouvant affecter jusqu’à 1 patient sur 10) :
· une augmentation du nombre d’infections qui peuvent vous causer maux de gorge, ulcères de la bouche ou fièvre, (cela peut indiquer que vous avez un faible taux de globules blancs (leucopénie, neutropénie).
Peu fréquent (pouvant affecter jusqu’à 1 patient sur 100) :
· réactions allergique telles qu’éruption cutanée, démangeaisons ou gonflement de la face, des lèvres, de la langue ou de la gorge, difficulté à avaler ou respirer ;
· caillots sanguins dans les veines, en particulier dans les jambes (les signes comprennent gonflement, douleur et rougeur de la jambe), qui peuvent se déplacer à travers les vaisseaux sanguins jusqu’aux poumons entraînant des douleurs de la poitrine et des difficultés à respirer ;
· apparition ou aggravation d’un diabète, occasionnellement associé à une acidocétose (corps cétoniques dans le sang et dans les urines occasionnant une perte d’appétit, une perte de poids inexpliquée, des nausées, des vomissements, des douleurs à l’estomac, des difficultés à respirer, un ralentissement du rythme cardiaque, des douleurs musculaires inhabituelles ou une sensation de faiblesse, de fatigue ou d’inconfort) ou un coma ;
· rythme cardiaque irrégulier ;
· convulsions, généralement associées à un antécédent de convulsions (épilepsie) ;
· mouvements incontrôlés de la bouche, de la langue, des joues ou des mâchoires qui peuvent évoluer vers les bras et les jambes (dyskinésie tardive) ;
· difficulté à uriner ou à vider la vessie.
Rare (pouvant affecter jusqu’à 1 patient sur 1 000) :
· jaunissement de votre peau et de la partie blanche de vos yeux, urine foncée, selles pâles, démangeaisons, sensation de somnolence ou de fatigue, fièvre, nausée, faiblesse et douleurs abdominales (qui peuvent être le signe d’un problème au foie) ;
· association d’une très forte fièvre, d’une respiration plus rapide, d’une sudation excessive, d’une modification de l’humeur, d’une raideur musculaire, d’une augmentation de la pression sanguine et d’une somnolence ou endormissement (syndrome malin des neuroleptiques) ;
· rythme cardiaque inhabituel ou dangereusement rapide (tachycardie ventriculaire/fibrillation) ;
· inflammation du pancréas entraînant des douleurs aiguës à l’estomac, qui se propagent jusqu’au dos ;
· diminution de la température corporelle causant frissons, peau froide ou pâle ;
· atteinte musculaire entraînant une douleur musculaire, une faiblesse ou des courbatures associées à des urines foncées (rhabdomyolyse) ;
· érection prolongée et/ou douloureuse.
Fréquence indéterminée (ne peut être déterminée sur la base des données disponibles) :
· symptômes évocateurs de la grippe et une éruption sur le visage, puis sur tout le corps, avec élévation de la température, gonflement des ganglions lymphatiques, accroissement des niveaux d'enzymes du foie dans le sang et augmentation d'un type de globules blancs (éosinophilie). Cela peut être le signe d’un syndrome d'hypersensibilité médicamenteuse (DRESS).
Autres effets indésirables possibles
Très fréquent (pouvant affecter plus d’1 patient sur 10) :
· prise de poids ;
· envie de dormir ;
· augmentation des taux de prolactine pouvant être détectée dans un test sanguin ;
· au début du traitement, certaines personnes peuvent éprouver des vertiges ou des sensations de malaise (avec un pouls ralenti), en particulier au moment de se mettre debout après avoir été allongé ou assis. Ces effets disparaissent habituellement spontanément, mais dans le cas contraire, veuillez-en informer votre médecin.
Fréquent (pouvant affecter jusqu’à 1 patient sur 10) :
· augmentations du taux de globules blancs, de lipides circulants et, au début du traitement, augmentation transitoire des enzymes du foie, qui peuvent être détectées par un test sanguin ;
· augmentation des taux de sucre dans le sang et l’urine, qui peuvent être détectées par un test sanguin ou urinaire ;
· augmentation des taux de l’acide urique, de la phosphatase alcaline et de la créatine phosphokinase qui peuvent être détectées par un test sanguin ;
· augmentation de la sensation de faim ;
· vertiges ;
· impatience ou difficultés à rester assis ;
· tremblements, rigidité, mouvements lents, démarche traînante et instable (Parkinsonisme) ;
· mouvements anormaux (dyskinésies) ;
· constipation ;
· bouche sèche ;
· éruption cutanée ;
· faiblesse inhabituelle ;
· fatigue intense ;
· rétention d’eau pouvant conduire à un gonflement au niveau des mains, des chevilles ou des pieds ;
· fièvre ;
· douleurs articulaires ;
· problèmes sexuels tels que diminution du désir sexuel chez les hommes et chez les femmes ou difficulté pour avoir ou maintenir une érection chez les hommes.
Peu fréquent (pouvant affecter jusqu’à 1 patient sur 100) :
· raideur ou spasmes musculaires incontrôlés au niveau de la tête (dont des mouvements des yeux), du cou et du corps ;
· syndrome des Jambes Sans Repos ;
· problèmes d’élocution ;
· bégaiement ;
· ralentissement du pouls ;
· augmentation de la sensibilité de la peau au soleil ;
· saignement de nez ;
· ballonnement (distension abdominale) ;
· excès de salive ;
· perte de mémoire ou moment d’inattention ;
· incapacité à contrôler la miction, difficulté à uriner ou à maintenir le jet urinaire ;
· perte de cheveux ;
· absence ou diminution des règles ;
· modification de la taille de la poitrine chez les hommes et chez les femmes ;
· production anormale de lait chez les femmes ;
· augmentation du taux de bilirubine dans le sang détectée par un test sanguin.
Rares (pouvant affecter jusqu’à 1 patient sur 1 000) :
· signes de sevrage tels que transpiration, troubles du sommeil, agitation, anxiété, sensation ou être malade ;
· saignements ou ecchymoses inexpliqués, plus facilement ou plus persistants que d’habitude (thrombocytopénie) ;
· mort soudaine inexpliquée.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
· signes de sevrage chez les nouveau-nés tels que peau irritée, diarrhée, succion ou pleurs excessifs, manque d’appétit, prise de poids lente, éternuement.
Lors de la prise d’olanzapine, les patients âgés souffrant de démence peuvent présenter un accident vasculaire cérébral, une pneumonie, une incapacité à contrôler la miction, des chutes, une extrême fatigue, des hallucinations visuelles (voir des choses qui n’existent pas), une augmentation de la température corporelle, une rougeur de la peau et des troubles de la marche. Dans ce groupe spécifique de patients, des décès ont été rapportés.
Chez les patients atteints de la maladie de Parkinson, l’olanzapine peut aggraver les symptômes et entraîner des hallucinations (voir, entendre ou sentir des choses qui n’existent pas).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER OLANZAPINE VIATRIS 5 mg, comprimé orodispersible ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte, la plaquette ou le flacon. La date de péremption fait référence au dernier jour de ce mois.
À conserver dans l’emballage d'origine à l’abri de la lumière et de l’humidité.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient OLANZAPINE VIATRIS 5 mg, comprimé orodispersible
· La substance active est : l'olanzapine.
Chaque comprimé orodispersible contient 5 mg d’olanzapine.
· Les autres composants sont :
mannitol, cellulose microcristalline, gomme guar, crospovidone, stéarate de magnésium, silice colloïdale anhydre, aspartame (E951, voir rubrique 2 « OLANZAPINE VIATRIS contient de l’aspartame ») et laurilsulfate de sodium.
Qu’est-ce que OLANZAPINE VIATRIS 5 mg, comprimé orodispersible et contenu de l’emballage extérieur
Ce médicament se présente sous la forme de comprimé jaune à jaune clair, uni à moucheté, rond, plat, à bords biseautés, gravé « M » sur une face et « OE1 » sur l'autre face.
OLANZAPINE VIATRIS comprimé orodispersible est proposé en plaquettes non prédécoupées contenant 7, 10, 14, 28, 30, 35, 56, 60, 70 98, 100 comprimés, en plaquettes prédécoupées pour dose unitaire contenant (7, 10, 14, 28, 30, 35, 56, 60, 70, 98 ou 100) x1 comprimés et en flacons contenant 7, 10, 14, 28, 30, 56, 98, 100, 250, 500 comprimés.
Les flacons contiennent un desséchant ; ne pas avaler le desséchant.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
VIATRIS SANTE
1 RUE DE TURIN
69007 LYON
Exploitant de l’autorisation de mise sur le marché
VIATRIS SANTE
1 RUE DE TURIN
69007 LYON
H-2900 KOMAROM
MYLAN UTCA 1
HONGRIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).