Dernière mise à jour le 01/06/2026
URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion
Indications thérapeutiques
Ce médicament est utilisé chez les adultes :
· en cas d'urgence hypertensive (par exemple, en cas d'élévation soudaine et importante de la pression artérielle appelée « crise hypertensive ») ;
· pour traiter les formes graves à extrêmement graves d'hypertension ou l'hypertension résistant au traitement ;
· pour réduire la pression artérielle pendant et/ou après une opération.
Présentations
> 5 ampoule(s) en verre de 10 ml
Code CIP : 34009 302 243 9 7
Déclaration de commercialisation : 22/02/2023
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 26/06/2024
URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour 1 mL.
Chaque ampoule de 10 mL contient 50 mg d’urapidil.
Excipients à effet notoire :
Ce médicament contient du propylène glycol (E1520).
1 mL de solution contient 100 mg de propylène glycol.
10 mL de solution contient 1000 mg de propylène glycol.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable/pour perfusion.
Solution limpide, incolore, exempte de particules visibles.
pH compris entre 5,6 et 6,6.
Osmolalité approximative de 1700 mOsmol/kg.
4.1. Indications thérapeutiques
Réduction contrôlée de la pression artérielle chez les patients hypertendus pendant et/ou après une intervention chirurgicale.
URAPIDIL KALCEKS est indiqué chez les adultes.
4.2. Posologie et mode d'administration
Posologie
Urgence hypertensive, formes graves et extrêmement graves d'hypertension et hypertension résistante aux traitements
1) Injection intraveineuse
En injection, 10 à 50 mg d'urapidil sont administrés lentement, avec une surveillance continue de la pression artérielle, par voie intraveineuse.
Un effet hypotenseur peut être attendu dans les 5 minutes suivant l'injection. En fonction de la réponse tensionnelle, l'injection d'urapidil peut être répétée.
2) Perfusion intraveineuse continue au goutte-à-goutte ou perfusion continue par pousse-seringue
La solution pour perfusion continue au goutte-à-goutte, utilisée pour maintenir le niveau de pression sanguine atteint avec l'injection, est préparée comme suit : 250 mg d'urapidil sont généralement ajoutés à 500 mL d'une solution pour perfusion compatible (voir rubrique 6.6).
Lorsqu’un pousse-seringue est utilisé pour administrer la dose d'entretien, 20 mL de solution injectable/pour perfusion (= 100 mg d'urapidil) sont aspirés dans un pousse-seringue et dilués à un volume de 50 mL avec une solution pour perfusion compatible (voir rubrique 6.6).
La quantité maximale compatible est de 4 mg d'urapidil par mL de solution pour perfusion.
Débit d'administration
Le débit de perfusion doit être basé sur la réponse individuelle de la pression artérielle.
Débit initial recommandé : 2 mg/min.
L'ampleur de la réduction de la pression artérielle est déterminée par la dose perfusée dans les 15 premières minutes. Par la suite, la pression artérielle établie peut être maintenue avec des doses significativement plus faibles.
Dose d'entretien : 9 mg/h en moyenne, sur la base de 250 mg d'urapidil dans 500 mL de solution pour perfusion, équivalent à 1 mg = 44 gouttes = 2,2 mL.
Réduction contrôlée de la pression artérielle en cas d'hypertension pendant et/ou après une intervention chirurgicale
Pour maintenir le niveau de pression artérielle atteint lors de l'injection, une perfusion continue via un pousse-seringue ou une perfusion continue au goutte-à-goutte est utilisée.
Régime posologique
|
Injection intraveineuse de 25 mg d’urapidil (= 5 mL de solution injectable/pour perfusion) |
si la pression artérielle diminue |
Pression artérielle stabilisée par perfusion
Initialement jusqu’à 6 mg pendant 1‑2 minutes, puis réduction de la dose |
|
|
après 2 min. |
|||
|
après 2 min. |
|
|
|
|
Injection intraveineuse de 25 mg d’urapidil (= 5 mL de solution injectable/pour perfusion) |
si la pression artérielle diminue |
||
|
après 2 min. |
|||
|
après 2 min. |
pas de réponse de la pression artérielle |
|
|
|
Injection intraveineuse lente de 50 mg d’urapidil (= 10 mL de solution injectable/pour perfusion) |
si la pression artérielle diminue |
||
|
après 2 min. |
|||
Populations particulières de patients
Posologie en cas d'insuffisance hépatique
Chez les patients souffrant de dysfonctionnement hépatique, il peut être nécessaire de réduire la dose d'urapidil.
Posologie en cas d'insuffisance rénale
Chez les patients souffrant de dysfonctionnement rénal, il peut être nécessaire de réduire la dose d'urapidil.
Personnes âgées
Chez les personnes âgées, les antihypertenseurs doivent être administrés avec prudence et à des doses plus faibles au début du traitement, car la sensibilité à ces médicaments est souvent altérée chez ces patients.
Population pédiatrique
La sécurité et l'efficacité de l'urapidil chez les enfants et les adolescents n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Voie intraveineuse.
URAPIDIL KALCEKS est administré par voie intraveineuse sous forme d'injection ou de perfusion aux patients placés en décubitus dorsal.
Des injections uniques et multiples, ainsi que des perfusions de longue durée, sont possibles. Les injections peuvent être associées à des perfusions ultérieures de longue durée.
En cas de chevauchement avec un traitement parentéral aigu, il est possible de passer à un traitement d'entretien avec des antihypertenseurs administrés par voie orale.
Pour éviter les effets toxicologiques, la durée de traitement ne doit pas dépasser 7 jours, ce qui est généralement le cas avec le traitement antihypertenseur parentéral. Le traitement parentéral peut être répété en cas de récidive de l'hypertension.
URAPIDIL KALCEKS ne doit pas être utilisé dans les cas suivants :
· Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1 ;
· Patients présentant une sténose de l'isthme aortique et un shunt artério-veineux (exception : shunts artério-veineux des hémodialysés).
· Allaitement maternel.
4.4. Mises en garde spéciales et précautions d'emploi
Avertissements
Une chute trop rapide de la pression artérielle peut entraîner une bradycardie ou un arrêt cardiaque.
Le « syndrome de l'iris flasque peropératoire » (IFIS, une variante du syndrome de pupille étroite) a été observé lors d'une opération de la cataracte chez certains patients traités ou précédemment traités par la tamsulosine. Des cas isolés ont également été rapportés avec d'autres alpha1-bloquants et la possibilité d'un effet de classe ne peut être exclue. L'IFIS pouvant entraîner une augmentation des complications procédurales pendant l'opération de la cataracte, l'utilisation actuelle ou passée des alpha1-bloquants doit être portée à la connaissance du chirurgien ophtalmologiste avant l'opération.
Précautions
Une prudence particulière est nécessaire avec l’utilisation d'urapidil dans les cas suivants :
· insuffisance cardiaque causée par une obstruction fonctionnelle mécanique, par exemple sténose de la valve aortique ou mitrale, embolie pulmonaire ou altération de la fonction cardiaque causée par une maladie péricardique ;
· patients souffrant d'un dysfonctionnement hépatique ;
· patients souffrant d'un dysfonctionnement rénal modéré à grave ;
· patients âgés ;
· patients recevant de la cimétidine de façon concomitante (voir rubrique 4.5).
Si d'autres agents antihypertenseurs ont été administrés auparavant, il convient de prévoir un délai suffisant pour que le ou les agents antihypertenseurs précédents fassent effet. Une dose plus faible d'urapidil doit être choisie en conséquence.
Excipients
URAPIDIL KALCEKS contient du propylène glycol (E1520)
Ce médicament contient du propylène glycol (voir rubrique 2), qui peut avoir les mêmes effets que l’absorption d'alcool et augmenter la probabilité d’effets indésirables.
Chez les femmes enceintes et les patients dont la fonction hépatique et/ou rénale est altérée, ce médicament ne peut être administré que sur recommandation d'un médecin. Pendant l'administration de ce médicament, une surveillance supplémentaire peut être effectuée sur les instructions d'un médecin.
URAPIDIL KALCEKS contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par mL de solution, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L'effet antihypertenseur de l'urapidil peut être potentialisé par la co-administration d’alpha-bloquants, de vasodilatateurs et d'autres agents antihypertenseurs, ainsi que dans les états de déplétion du volume (diarrhées, vomissements) et par l'alcool.
Une augmentation de 15 % des taux sériques d'urapidil peut être attendue si la cimétidine est coadministrée.
Comme il n'existe pas encore d'expérience adéquate en ce qui concerne le traitement associé avec des inhibiteurs de l'ECA, un tel traitement n'est actuellement pas recommandé.
L'urapidil à fortes doses peut prolonger la durée d'action des barbituriques.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer
Ce médicament n'est pas recommandé aux femmes en âge de procréer qui n’utilisent pas de contraception.
Grossesse
Il n'existe pas encore de données ou il existe des données très limitées sur l'utilisation de l'urapidil chez les femmes enceintes.
Des études sur les animaux ont montré une toxicité sur la reproduction (voir rubrique 5.3). L'urapidil traverse le placenta.
Ce médicament ne doit pas être utilisé pendant la grossesse, sauf si un traitement à l'urapidil est nécessaire en raison de l'état clinique de la patiente.
Allaitement
On ne sait pas si l'urapidil est excrété dans le lait maternel. Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Ce médicament ne doit pas être utilisé pendant l'allaitement.
Fertilité
Aucune étude clinique n'a été conduite pour évaluer l’effet de l’urapidil sur la fertilité masculine et féminine.
Les études effectuées chez l’animal ont montré que l'urapidil a un effet sur la fertilité (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Dans la majorité des cas, les effets indésirables suivants sont attribuables à une chute excessivement rapide de la pression artérielle. Cependant, l'expérience clinique a montré qu'ils disparaissent en quelques minutes, même lors de perfusions de longue durée. Par conséquent, la décision d'interrompre le traitement doit être prise en fonction de la gravité des effets indésirables.
Les catégories suivantes sont utilisées pour exprimer la fréquence des effets indésirables :
· Fréquents (≥ 1/100 à < 1/10)
· Peu fréquent (≥ 1/1 000 à < 1/100)
· Rare (≥ 1/10 000 à < 1/1 000)
· Très rare (< 1/10 000)
· Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
|
Fréquence
Système de classe organe |
Fréquent |
Peu fréquent |
Rare |
Très rare |
Fréquence indéterminée |
|
Affections psychiatriques |
|
Troubles du sommeil
|
|
Agitation |
|
|
Affections du système nerveux |
Vertiges, céphalées |
|
|
|
|
|
Affections cardiaques |
|
Palpitations, tachycardie, bradycardie, sensation d’oppression ou douleur thoracique (similaire à l’angor), dyspnée |
|
|
|
|
Affections vasculaires |
|
Hypotension posturale (dysrégulation orthostatique) |
|
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Congestion nasale |
|
|
|
Affections gastro-intestinales |
Nausées |
Vomissements, diarrhée, sécheresse de la bouche |
|
|
|
|
Affections de la peau et du tissu sous-cutané |
|
Episode d’hyperhidrose |
Hypersensibilité telle que prurit, érythème, exanthème |
|
Angiœdème, urticaire |
|
Affection du rein et des voies urinaires |
|
|
|
Miction impérieuse, aggravation d’une l’incontinence urinaire |
|
|
Affections des organes de reproduction et du sein |
|
|
Priapisme |
|
|
|
Troubles généraux et anomalies au site d’administration |
|
Fatigue |
|
|
|
|
Investigations |
|
Rythme cardiaque irrégulier |
|
Diminution de la numération plaquettaire* |
|
* Dans de très rares cas isolés, une réduction du nombre de plaquettes a été observée pendant l’administration orale d’urapidil. Aucune relation de causalité avec le traitement par l’urapidil n’a été établie, par exemple, par des tests immuno-hématologiques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Symptômes d'un surdosage
Système circulatoire : vertiges, hypotension orthostatique, collapsus.
Système nerveux central : fatigue et diminution de la réactivité.
Mesures de traitement des surdosages
Une baisse excessive de la pression artérielle peut être corrigée en surélevant les jambes et en effectuant un remplissage vasculaire. Si ces mesures sont insuffisantes, des vasoconstricteurs peuvent être administrés par injection intraveineuse lente sous surveillance de la pression artérielle. Dans de très rares cas, l'administration de catécholamines est nécessaire (par exemple, adrénaline 0,5-1,0 mg diluée à 10 mL avec une solution isotonique de chlorure de sodium).
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d'action
L'Urapidil entraîne une réduction de la pression artérielle systolique et diastolique, en diminuant la résistance périphérique.
Le rythme cardiaque reste en grande partie constant.
Le débit cardiaque n'est pas altéré ; les cas de réduction du débit cardiaque due à une augmentation de la postcharge peuvent augmenter.
L'urapidil est un vasodilatateur ayant des effets centraux et périphériques.
Sur le plan périphérique, l'urapidil bloque principalement les récepteurs alpha1-adrénergiques post-synaptiques, inhibant ainsi l'effet vasoconstricteur des catécholamines.
Au niveau central, l'urapidil module l'activité des centres de régulation circulatoire, ce qui empêche une augmentation réflexe du tonus sympathique ou réduit le tonus sympathique. L'urapidil régule la pression artérielle et le tonus sympathique par une inhibition de l'activité des récepteurs α1 adrénergiques et une stimulation des récepteurs sérotoninergiques 5-HT1A.
5.2. Propriétés pharmacocinétiques
Absorption
Après l'administration intraveineuse de 25 mg d'urapidil, la progression biphasique (phase de distribution initiale, phase d'élimination terminale) des concentrations sanguines a été mesurée.
La phase de distribution a une demi-vie d'environ 35 minutes. Le volume de distribution est de 0,8 (0,6-1,2) litres/kg.
Pour la demi-vie d'élimination dans le plasma, 2,7 (1,8-3,9) heures ont été mesurées après l'injection du bolus intraveineux.
La liaison in vitro de l'urapidil aux protéines plasmatiques (sérum humain) est de 80 %. Cette liaison relativement faible de l'urapidil aux protéines plasmatiques pourrait expliquer qu'à ce jour, il n'existe aucune interaction connue entre l'urapidil et les médicaments à forte liaison aux protéines plasmatiques.
Distribution
Le volume de distribution est de 0,77 litres/kg de poids corporel. La substance pénètre la barrière hémato-encéphalique et traverse le placenta.
Biotransformation
L'urapidil est principalement métabolisé dans le foie. Le principal métabolite est l'urapidil hydroxylé sur en position 4 du noyau phényle, qui n'a pas d'effet antihypertensif notable. Le métabolite urapidil O-déméthylé possède globalement la même activité biologique que l'urapidil, mais n'est produit que dans une moindre mesure.
Élimination
Chez l'Homme, environ 50 à 70 % de l'urapidil et de ses métabolites sont éliminés par voie rénale, dont environ 15 % de la dose administrée sous forme d'urapidil pharmacologiquement actif. Le reste est excrété sous forme de métabolites avec les fèces, principalement sous forme d'urapidil parahydroxylé non hypotenseur.
Populations particulières de patients
En cas d'insuffisance hépatique et/ou rénale avancée, ainsi que chez les patients âgés, le volume de distribution et la clairance de l'urapidil sont réduits et la demi-vie d'élimination est prolongée.
5.3. Données de sécurité préclinique
Toxicité pour la reproduction et le développement
Dans les études de toxicité sur la reproduction sur les souris, les rats et les lapins, aucun résultat tératogène lié à l'urapidil n'a été identifié.
Dans les études de toxicité chronique et de toxicité sur la reproduction chez les rats et les souris, des effets sur la fertilité des mâles ont été observés, ainsi que des effets histopathologiques sur les organes reproducteurs des femelles.
L’allongement ou l’absence du cycle œstral observé chez les rats femelles ainsi que la réduction du poids de l'utérus sont attribués à l'augmentation du taux de prolactine induite par le traitement à l'urapidil et étaient réversibles après l’arrêt du traitement. La fertilité des femelles n'a pas été affectée. La pertinence de ces observations pour l'Homme n’est pas connue en raison des différences entre espèces. Aucun effet sur le système hypophyso-gonadique n'a été observé chez les femmes dans les études cliniques à long terme.
Dans les études sur le développement embryo-fœtal chez le lapin, une augmentation du taux de mortalité fœtale a été observée ainsi qu'une toxicité maternelle concomitante.
Dans les études péri et postnatales sur les rats, une augmentation de la mortalité fœtale due à l'urapidil et une réduction du poids à la naissance a été observée dans la génération F1. La génération F2 n'a fait l'objet d'aucun résultat.
Aucune donnée toxicocinétique (Cmax, ASC) n'a été présentée. Les marges de sécurité concernant l'exposition clinique ne peuvent donc pas être estimées.
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l’exception de ceux mentionnés dans la rubrique 6.6.
3 ans.
Durée de conservation après dilution
La stabilité physico-chimique en cours d’utilisation a été démontrée pendant 50 heures à 25°C et à 2‑8°C lorsque la solution est diluée dans 9 mg/mL (0,9 %) de chlorure de sodium ou 50 mg/mL (5 %) de glucose, ou 100 mg/mL (10 %) de solution de glucose pour perfusion.
D'un point de vue microbiologique, la solution diluée doit être utilisée immédiatement. En cas d’utilisation non immédiate, la durée et les conditions de conservation avant utilisation sont de la responsabilité de l'utilisateur et ne devraient normalement pas dépasser 24 heures entre 2 et 8°C, à moins que la dilution n'ait eu lieu dans des conditions d’asepsie contrôlées et validées.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
Pour les conditions de conservation du médicament après dilution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Ampoules en verre transparent de type I de 10 mL avec un système d’ouverture OPC (One Point Cut).
5 ampoules sont emballées dans une pellicule. La pellicule est emballée dans une boîte.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
A usage unique.
Le médicament doit être utilisé immédiatement après l'ouverture de l'ampoule. Toute solution non utilisée doit être jetée.
Le médicament doit être inspecté visuellement avant utilisation. Seules des solutions claires et exemptes de particules doivent être utilisées.
Peut être dilué avec :
· 9 mg/mL (0,9 %) de solution de chlorure de sodium pour perfusion ;
· 50 mg/mL (5 %) de solution de glucose pour perfusion ;
· 100 mg/mL (10 %) de solution de glucose pour perfusion.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
AS KALCEKS
KRUSTPILS IELA 71E
RĪGA, LV‑1057
LETTONIE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 243 9 7 : 10 mL en ampoule (verre). Boîte de 5.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 26/06/2024
URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion
Urapidil
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre infirmier/ère.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d’utiliser URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion ?
3. Comment utiliser URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Ce médicament est utilisé chez les adultes :
· en cas d'urgence hypertensive (par exemple, en cas d'élévation soudaine et importante de la pression artérielle appelée « crise hypertensive ») ;
· pour traiter les formes graves à extrêmement graves d'hypertension ou l'hypertension résistant au traitement ;
· pour réduire la pression artérielle pendant et/ou après une opération.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT d’utiliser URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion ?
Vous ne devez jamais recevoir URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion :
· si vous êtes allergique à l'urapidil ou à l'un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· si vous avez un rétrécissement de l'artère principale (sténose aortique) ou une anomalie des vaisseaux sanguins appelée « shunt cardiaque » (sauf le shunt cardiaque chez les personnes sous dialyse) ;
· si vous allaitez.
Avertissements et précautions
Une baisse de la fréquence cardiaque ou un arrêt cardiaque peut se produire si la pression artérielle baisse trop rapidement.
Adressez-vous à votre médecin ou votre infirmier/ère avant de recevoir ce médicament si l'une des situations suivantes s'applique à vous, car une prudence particulière est requise :
· vous avez eu des diarrhées ou des vomissements (ou toute autre cause de réduction des liquides dans votre corps) ;
· chez les patients souffrant d'une insuffisance cardiaque causée par une obstruction mécanique, par exemple un rétrécissement des valves cardiaques (sténose de la valve aortique ou mitrale) ;
· chez les patients présentant une obstruction d'une artère des poumons (embolie pulmonaire) ;
· chez les patients dont la fonction cardiaque est altérée par une inflammation du sac tissulaire qui entoure le cœur (maladie péricardique) ;
· chez les patients souffrant de troubles hépatiques ;
· chez les patients souffrant de troubles rénaux modérés à graves ;
· chez les personnes âgées ;
· chez les patients utilisant simultanément de la cimétidine (médicament pour réduire l'acide gastrique).
Si vous n'êtes pas sûr que l'une des situations ci-dessus s'applique à vous, parlez-en à votre médecin ou à votre infirmier/ère.
Si vous devez subir une opération des yeux en raison d'une cataracte (opacification du cristallin), veuillez informer votre ophtalmologue avant l'opération que vous utilisez ou avez déjà utilisé de l'urapidil. En effet, l'urapidil peut entraîner des complications pendant l'opération, qui peuvent être prises en charge si votre spécialiste est préparé à l'avance.
Si un autre médicament pour abaisser la tension artérielle a été administré avant l'urapidil, le médecin attendra suffisamment longtemps pour que le médicament administré précédemment soit efficace. Votre médecin réduira la dose d'urapidil. Une baisse trop rapide de la pression artérielle peut entraîner une diminution du rythme cardiaque ou un arrêt cardiaque.
Enfants et adolescents
Ce médicament ne doit pas être utilisé chez les enfants et les adolescents.
Autres médicaments et URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion
Informez votre médecin ou votre infirmier/ère si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Informez votre médecin ou votre infirmier/ère avant de recevoir ce médicament si vous prenez l'un des médicaments suivants, car ils peuvent interagir avec URAPIDIL KALCEKS en modifiant les effets des médicaments ou en rendant les effets secondaires plus probables :
· alpha-bloquants (médicaments utilisés pour traiter les troubles des voies urinaires associés à une maladie de la prostate) ;
· tout médicament destiné à diminuer la pression artérielle ;
· cimétidine (utilisée pour inhiber la production d'acide gastrique) ;
· barbituriques (médicaments utilisés pour traiter l'épilepsie).
URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion avec de l’alcool
L'alcool peut augmenter l'effet de ce médicament.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de recevoir ce médicament.
Il n'existe pas de données adéquates pour déterminer la sécurité de l'urapidil chez les femmes enceintes. Ce médicament ne doit pas être utilisé pendant la grossesse, sauf si l'état clinique de la patiente l'exige. Si une hypertension artérielle survient pendant une grossesse et doit être traitée avec ce médicament, la réduction de la pression artérielle doit être progressive et toujours surveillée par un médecin.
On ne sait pas si ce médicament est excrété dans le lait maternel. Pour des raisons de sécurité, ce médicament ne doit pas être utilisé pendant l'allaitement.
Ce médicament est déconseillé aux femmes en âge de procréer qui n’utilisent pas de contraception.
Des études sur les animaux ont montré que l'urapidil affecte la fertilité. Toutefois, l'importance de cet effet chez l'Homme n’est pas connue.
Conduite de véhicules et utilisation de machines
Ce médicament peut affecter votre capacité à conduire ou à faire fonctionner des machines, plus particulièrement au début du traitement, en cas d'augmentation de la dose ou de changement de médicament, ou en association avec l'alcool.
URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion contient du propylène glycol (E1520) et du sodium
Propylène glycol :
· Ce médicament contient 1000 mg de propylène glycol pour 10 mL de solution équivalent à 100 mg/mL.
· Si vous êtes enceinte ou si vous allaitez, ne prenez pas ce médicament sauf avis contraire de votre médecin. Votre médecin pourra procéder à des contrôles supplémentaires pendant que vous prenez ce médicament.
· Si vous souffrez d’une maladie du foie ou du rein, ne prenez ce médicament que sur avis de votre médecin. Votre médecin pourra procéder à des examens complémentaires pendant que vous prenez ce médicament.
· Le propylène glycol contenu dans ce médicament peut avoir les mêmes effets que l’absorption d’alcool et augmenter la probabilité d’effets indésirables.
Sodium :
Ce médicament contient moins de 1 mmol (23 mg) de sodium par mL, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion ?
Comment ce médicament est-il administré ?
· Ce médicament sera administré par un professionnel de santé.
· Ce médicament sera administré par injection ou perfusion dans une veine. Il peut être administré sous forme d'injections uniques ou répétées, ou en perfusions de longue durée. Les injections peuvent être associées (suivies) à des perfusions de longue durée.
· Vous devez être en position couchée pendant l'administration de ce médicament.
· Pendant le traitement, votre pression artérielle sera constamment surveillée.
Dosage
Le médecin décidera du dosage approprié en fonction de votre état.
Crise hypertensive et formes graves à extrêmement graves d'hypertension ou d'hypertension résistant au traitement
Par injection dans une veine
En injection, 10 à 50 mg d'urapidil sont administrés lentement, avec une surveillance continue de la pression artérielle. L’effet de diminution de la pression artérielle peut être attendu dans les 5 minutes suivant l’injection. En fonction de la réponse de la pression artérielle, l'injection d'urapidil peut être répétée.
Par perfusion dans une veine (au goutte-à-goutte ou à l'aide d'un pousse-seringue)
Pour une perfusion au goutte-à-goutte continue, 250 mg d'urapidil sont ajoutés à 500 mL d'une solution pour perfusion compatible (solution de chlorure de sodium à 0,9 %, ou de glucose à 5 % ou 10 %).
Lorsqu’un pousse-seringue est utilisé, 100 mg d'urapidil sont aspirés dans un pousse-seringue et dilués à un volume de 50 mL avec une solution pour perfusion compatible (voir ci-dessus) (maximum 4 mg d'urapidil par mL de solution pour perfusion).
La vitesse de perfusion initiale est de 2 mg/min. La dose d'entretien est en moyenne de 9 mg/heure. La diminution de la pression artérielle sera déterminée par la dose perfusée dans les 15 premières minutes. La pression artérielle établie peut ensuite être maintenue avec des doses significativement plus faibles.
Réduction de la pression artérielle pendant et/ou après une intervention chirurgicale
Pour maintenir le niveau de pression sanguine atteint lors de l'injection, une perfusion continue via un pousse-seringue ou une perfusion continue au goutte-à-goutte est utilisée.
Par injection dans une veine
Dans un premier temps, 25 mg d'urapidil sont administrés. Cette dose sera répétée si aucune baisse suffisante de la pression artérielle n'est obtenue après 2 minutes. Si, dans les 2 minutes suivant la deuxième dose, la baisse de la pression artérielle est toujours insuffisante, 50 mg d'urapidil seront administrés.
Si la baisse de la pression artérielle après 2 minutes suivant l'administration de la dose est suffisante, vous passerez à la dose d'entretien.
Par perfusion dans une veine (au goutte-à-goutte ou à l'aide d'un pousse-seringue)
Dans un premier temps, jusqu'à 6 mg pendant 1-2 minutes seront administrés. Ensuite, la dose sera réduite.
Populations particulières de patients
Chez les patients souffrant de troubles hépatiques et/ou rénaux, il peut être nécessaire de réduire la dose.
Chez les personnes âgées, ce médicament doit être administré avec prudence. Des doses plus faibles seront administrées au début du traitement, car la sensibilité à ces médicaments est souvent altérée chez ces patients.
Durée du traitement
La durée du traitement avec ce médicament ne doit pas dépasser 7 jours.
Si vous avez reçu plus d’URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion que vous n’auriez dû
Si vous avez reçu plus de ce médicament que vous n’auriez dû, vous pouvez présenter des vertiges, des étourdissements ou des évanouissements lorsque vous vous mettez debout, de la fatigue et une vitesse de réaction plus lente. Dans ce cas, allongez-vous sur le dos avec les jambes surélevées. Si les symptômes persistent, informez immédiatement votre médecin ou votre infirmier/ère.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets secondaires décrits ci-dessous sont généralement la conséquence d'une chute trop soudaine de la pression artérielle.
Cependant, l'expérience a montré qu'ils disparaissent en quelques minutes, même lors d'une perfusion de longue durée. Le médecin décidera d'interrompre ou non le traitement en fonction de la gravité des effets secondaires.
Fréquents (pouvant affecter jusqu'à 1 personne sur 10) :
Étourdissements, maux de tête, nausées.
Peu fréquent (pouvant affecter jusqu'à 1 personne sur 100) :
Troubles du sommeil, palpitations, augmentation ou diminution du rythme cardiaque, sensation de d’oppression ou de douleur dans la poitrine (comme l'angine de poitrine), difficultés respiratoires, baisse de la pression artérielle en se levant d'une position assise ou couchée (dysrégulation orthostatique), vomissements, diarrhée, sécheresse de la bouche, transpiration, fatigue, rythme cardiaque irrégulier.
Rare (pouvant affecter jusqu'à 1 personne sur 1 000) :
Congestion nasale, réactions allergiques (démangeaisons, rougeurs de la peau, éruptions cutanées), érection prolongée et douloureuse.
Très rare (pouvant affecter jusqu'à 1 personne sur 10 000) :
Agitation, augmentation de l'envie d'uriner, augmentation de l'incontinence urinaire, diminution du nombre de plaquettes (cellules sanguines qui aident le corps à former des caillots pour arrêter les saignements).
Fréquence inconnue (ne peut être estimée sur la base des données disponibles) :
Urticaire, réaction allergique grave avec gonflement du visage, des lèvres, de la langue et de la gorge.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Après dilution
La stabilité physico-chimique en cours d’utilisation a été démontrée pendant 50 heures à 25°C et à 2‑8°C lorsque la solution est diluée dans 9 mg/mL (0,9 %) de chlorure de sodium ou 50 mg/mL (5 %) de glucose, ou 100 mg/mL (10 %) de solution de glucose pour perfusion.
D'un point de vue microbiologique, la solution diluée doit être utilisée immédiatement. En cas d’utilisation non immédiate, la durée et les conditions de conservation avant utilisation sont de la responsabilité de l'utilisateur et ne devraient normalement pas dépasser 24 heures entre 2 et 8°C, à moins que la dilution n'ait eu lieu dans des conditions d’asepsie contrôlées et validées.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et l’ampoule après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient URAPIDIL KALCEKS 50 mg/10 mL, solution injectable/pour perfusion
· La substance active est :
Urapidil............................................................................................................................. 5 mg
Pour 1 mL.
Chaque ampoule de 10 mL contient 50 mg d’urapidil.
· Les autres composants sont :
Acide chlorhydrique concentré, phosphate monosodique dihydraté, phosphate disodique dihydraté, propylène glycol (E1520), hydroxyde de sodium (pour l'ajustement du pH), eau pour préparations injectables.
Solution limpide, incolore, exempte de particules visibles.
10 mL en ampoule en verre transparent avec un système d’ouverture OPC (One Point Cut).
5 ampoules sont emballées dans une pellicule. La pellicule est emballée dans une boîte.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
KRUSTPILS IELA 71E
RĪGA, LV‑1057
LETTONIE
Exploitant de l’autorisation de mise sur le marché
84 QUAI JOSEPH GILLET
69004 LYON
KRUSTPILS IELA 71E
RĪGA, LV‑1057
LETTONIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
----------------------------------------------------------------------------------------------------------------------------------------
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Posologie
Urgence hypertensive, formes graves et extrêmement graves d'hypertension et hypertension résistante aux traitements
· Injection intraveineuse
En injection, 10 à 50 mg d'urapidil sont administrés lentement, avec une surveillance continue de la pression artérielle, par voie intraveineuse.
Un effet hypotenseur peut être attendu dans les 5 minutes suivant l'injection. En fonction de la réponse tensionnelle, l'injection d'urapidil peut être répétée.
· Perfusion intraveineuse continue au goutte-à-goutte ou perfusion continue par pousse-seringue
La perfusion continue en goutte-à-goutte ou le pousse-seringue sont utilisés pour maintenir le niveau de pression artérielle atteint avec l'injection. Pour les instructions de dilution de la solution, voir « Instructions pour l’utilisation et l’élimination » et « Préparation de la solution diluée » ci-dessous.
La quantité maximale compatible est de 4 mg d'urapidil par mL de solution pour perfusion.
Débit d'administration
Le débit de perfusion doit être basé sur la réponse individuelle de la pression artérielle.
Débit initial recommandé : 2 mg/min.
L'ampleur de la réduction de la pression artérielle est déterminée par la dose perfusée dans les 15 premières minutes. Par la suite, la pression artérielle établie peut être maintenue à des doses significativement plus faibles.
Dose d'entretien : 9 mg/h en moyenne, sur la base de 250 mg d'urapidil dans 500 mL de solution pour perfusion, équivalent à 1 mg = 44 gouttes = 2,2 mL.
Réduction contrôlée de la pression artérielle en cas d'hypertension pendant et/ou après une intervention chirurgicale
Pour maintenir le niveau de pression artérielle atteint lors de l'injection, une perfusion continue via un pousse-seringue ou une perfusion continue au goutte à goutte est utilisée.
Régime posologique
|
Injection intraveineuse de 25 mg d’urapidil (= 5 mL de solution injectable/pour perfusion) |
si la pression artérielle diminue |
Pression artérielle stabilisée par perfusion
Initialement jusqu’à 6 mg pendant 1‑2 minutes, puis réduction de la dose |
|
|
après 2 min. |
|||
|
après 2 min. |
|
|
|
|
Injection intraveineuse de 25 mg d’urapidil (= 5 mL de solution injectable/pour perfusion) |
si la pression artérielle diminue |
||
|
après 2 min. |
|||
|
après 2 min. |
pas de réponse de la pression artérielle |
|
|
|
Injection intraveineuse lente de 50 mg d’urapidil (= 10 mL de solution injectable/pour perfusion) |
si la pression artérielle diminue |
||
|
après 2 min. |
|||
Populations particulières de patients
Chez les patients souffrant de dysfonctionnement hépatique et/ou rénal, il peut être nécessaire de réduire la dose d'urapidil. Chez les personnes âgées, les antihypertenseurs doivent être administrés avec prudence et à des doses plus faibles au début du traitement, car la sensibilité à ces médicaments est souvent altérée chez ces patients.
Population pédiatrique
La sécurité et l'efficacité de l'urapidil chez les enfants et les adolescents n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Voie intraveineuse.
URAPIDIL KALCEKS est administré par voie intraveineuse sous forme d'injection ou de perfusion aux patients placés en décubitus dorsal.
Des injections uniques et multiples, ainsi que des perfusions de longue durée, sont possibles. Les injections peuvent être associées à des perfusions ultérieures de longue durée.
En cas de chevauchement avec un traitement parentéral aigu, il est possible de passer à un traitement d'entretien avec des antihypertenseurs administrés par voie orale.
Pour éviter les effets toxicologiques, la durée de traitement ne doit pas dépasser 7 jours, ce qui est généralement le cas avec le traitement antihypertenseur parentéral. Le traitement parentéral peut être répété en cas de récidive de l'hypertension.
Incompatibilités
Ce médicament ne doit pas être mélangé avec des solutions alcalines injectable ou pour perfusion, car les propriétés acides de la solution peuvent provoquer une turbidité ou une floculation.
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l’exception de ceux mentionnés ci-dessous.
Instructions pour l’utilisation et l’élimination
A usage unique.
A utiliser immédiatement après l'ouverture de l'ampoule. Toute solution non utilisée doit être jetée.
Le médicament doit être inspecté visuellement avant utilisation. Seules des solutions claires et exemptes de particules doivent être utilisées.
Préparation de la solution diluée
· Perfusion intraveineuse au goutte-à-goutte : ajouter 250 mg d’urapidil à 500 mL de l’une des solutions pour perfusion compatibles (voir ci-dessous).
· Pousse-seringue : 20 mL de solution injectable/pour perfusion (= 100 mg d’urapidil) sont aspirés dans un pousse-seringue et dilués à un volume de 50 mL avec une solution pour perfusion compatible (voir ci-dessous).
Peut être dilué avec :
· 9 mg/mL (0,9 %) de solution de chlorure de sodium pour perfusion ;
· 50 mg/mL (5 %) de solution de glucose pour perfusion ;
· 100 mg/mL (10 %) de solution de glucose pour perfusion.
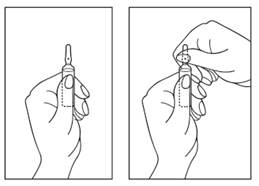
Instructions pour l’ouverture de l’ampoule
1) Tourner l’ampoule avec la pointe colorée vers le haut. S'il y a de la solution dans la partie supérieure de l'ampoule, tapotez doucement avec votre doigt pour faire passer toute la solution dans la partie inférieure de l'ampoule.
2) Utilisez les deux mains pour ouvrir ; tout en tenant la partie inférieure de l'ampoule d'une main, utilisez l'autre main pour casser la partie supérieure de l'ampoule dans la direction opposée au point coloré (voir les images ci-dessous).
|
|
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.