Dernière mise à jour le 29/06/2026
TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable
Indications thérapeutiques
Classe pharmacothérapeutique : ANTIDIARRHEIQUE ANTISECRETOIRE INTESTINAL. Code ATC : A07XA04. (A: appareil digestif et métabolisme).
TIORFAN 4 mg/mL suspension buvable est un médicament pour le traitement de la diarrhée.
TIORFAN 4 mg/mL suspension buvable est utilisé en complément de la réhydratation orale et des mesures diététiques dans le traitement des symptômes de la diarrhée aiguë du nourrisson et de l'enfant de plus de 3 mois et pesant 7 kg et plus. Il doit être pris avec autant de liquides qu’ils pourront boire et en association avec les mesures diététiques habituelles lorsque celles-ci ne suffisent pas à elles seules à maîtriser la diarrhée et si on ne peut pas remédier à la cause de la diarrhée.
Lorsque le traitement de la cause est possible, le racécadotril peut être administré en complément de ce traitement.
Présentations
> 1 flacon polytéréphtalate (PET) avec fermeture de sécurité enfant de 50 mL avec seringue de 10 mL graduée en kg
Code CIP : 34009 301 738 4 8
Déclaration de commercialisation : 12/10/2020
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 6,17 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 7,19 €
- Taux de remboursement :30%
> 1 flacon polytéréphtalate (PET) avec fermeture de sécurité enfant de 180 mL avec seringue de 10 mL graduée en kg
Code CIP : 34009 301 738 5 5
Déclaration de commercialisation : 12/10/2020
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 6,17 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 7,19 €
- Taux de remboursement :30%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Modéré | Avis du 11/03/2020 | Inscription (CT) | Le service médical rendu par TIORFAN 4 mg/ml nourrissons et enfants (racécadotril), solution buvable est modéré dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 11/03/2020 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux présentations déjà inscrites. |
ANSM - Mis à jour le : 17/04/2026
TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Racécadotril............................................................................................................................ 4 mg
Pour 1 mL de suspension buvable
Le flacon de 50 mL contient 168 mg de racécadotril, correspondant à 112 doses-kg
Le flacon de 180 mL contient 660 mg de racécadotril, correspondant à 440 doses-kg.
Chaque dose-kg correspond à 1,5 mg/kg/dose.
Excipients à effet notoire :
Chaque dose-kg de suspension buvable contient : 1,13 mg de benzoate de sodium, 0,84 mg de sodium, 225 mg de saccharose et 1,06 mg de propylène glycol.
Pour la liste complète des excipients, voir rubrique 6.1.
Suspension buvable.
Suspension de couleur blanche à blanc jaune orangé.
4.1. Indications thérapeutiques
TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS est indiqué en complément de la réhydratation orale et des mesures diététiques dans le traitement symptomatique des diarrhées aiguës du nourrisson et de l’enfant de plus de 3 mois et pesant 7 kg et plus, lorsque la réhydratation orale et les mesures diététiques seules ne suffisent pas à contrôler l'état clinique et lorsqu'un traitement causal n'est pas possible.
Si un traitement causal est possible, le racécadotril peut être administré en traitement complémentaire.
4.2. Posologie et mode d'administration
TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS est administré par voie orale en association avec une réhydratation orale (voir rubrique 4.4).
Posologie
Population pédiatrique
Réservé au nourrisson et à l’enfant de plus de 3 mois et pesant de 7 kg à 52 kg
La posologie usuelle est établie en fonction du poids corporel de l’enfant. Elle est de 1,5 mg/kg/prise (qui correspond à une dose-kg).
Le premier jour : une première prise d'emblée puis selon l’heure de la première prise, jusqu’à un maximum de 3 prises réparties dans la journée, en comptant dans ces 3 prises la première prise d’emblée. Les prises doivent se faire de préférence au début des trois principaux repas.
Les jours suivants : 3 prises réparties dans la journée, de préférence au début des trois principaux repas.
La posologie journalière maximale est de 3 prises.
Le médicament s’administre au moyen d’une seringue pour administration orale (graduée en kg de poids corporel) qui délivre une dose de 1,5 mg de racécadotril par graduation indiquée en kg.
Pour chaque prise :
· Nourrissons et enfants jusqu’à 26 kg : remplir la seringue jusqu’à la graduation indiquant le poids de l’enfant.
· Enfants entre 27 kg et 38 kg : remplir une première fois la seringue jusqu’à la graduation 13 kg et donner la suspension à l’enfant. Puis remplir une deuxième fois la seringue jusqu’à atteindre un total égal au poids de l’enfant et donner à nouveau la suspension à l’enfant.
· Enfants entre 39 kg et 52 kg : remplir une première fois la seringue jusqu’à la graduation 26 kg et donner la suspension à l’enfant. Puis remplir une deuxième fois la seringue jusqu’à atteindre un total égal au poids de l’enfant et donner à nouveau la suspension à l’enfant.
· Au-delà de 52 kg, il convient d’utiliser des formes pharmaceutiques plus adaptées.
Durée du traitement
Le traitement sera poursuivi jusqu'au retour de deux selles moulées consécutives, sans dépasser 7 jours.
Aucune étude clinique n'a été menée chez les nourrissons de moins de 3 mois.
Mode d’administration
Voie orale.
1) Agiter vigoureusement le flacon pour homogénéiser la suspension avant l’emploi
2) Ouvrir le flacon en tournant et en appuyant sur le bouchon sécurité-enfant
3) Introduire à fond la seringue dans l’embout de prélèvement
4) Pour remplir la seringue, tenir le flacon « tête en bas ». Bien maintenir la seringue en place et tirer doucement et régulièrement le piston jusqu’à la graduation nécessaire en kg
5) Remettre le flacon « tête en haut » et retirer la seringue
6) Introduire la seringue dans la bouche de l’enfant sans enfoncer et administrer la totalité de la suspension en appuyant doucement et progressivement sur le piston.
Après chaque utilisation, démonter la seringue pour administration orale, la rincer à l’eau et la sécher. L’usage de cette seringue pour administration orale est strictement réservé à l’administration de TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS.
Populations particulières :
Aucune étude n’a été menée chez les enfants souffrant d’insuffisance hépatique ou rénale (voir rubrique 4.4).
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Le traitement par TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS est un traitement adjuvant, à la réhydratation orale et ne dispense en aucun cas de celle-ci. La réhydratation doit être systématique chez les nourrissons/enfants présentant une diarrhée aiguë, afin de prévenir ou traiter la déshydratation, et doit être adaptée de façon à compenser les pertes hydroélectrolytiques.
Le traitement des diarrhées aiguës chez les nourrissons/enfants repose essentiellement sur la correction des pertes en eau et électrolytes par utilisation de solutés de réhydratation orale et la réalimentation précoce dont les modalités dépendent de l'âge de l'enfant et du type de l'alimentation antérieure à la diarrhée.
L'importance de la réhydratation par soluté de réhydratation orale ou par voie intraveineuse doit être adaptée en fonction de l'intensité de la diarrhée, de l'âge de l'enfant et des maladies associées.
En cas de diarrhée sévère ou prolongée, de vomissement important ou de refus d'alimentation, une réhydratation par voie intraveineuse devra être envisagée.
La présence de sang ou de pus dans les selles avec de la fièvre peut être le signe d'une diarrhée infectieuse ou de la présence d'autres maladies en cours. En cas de diarrhée infectieuse avec des manifestations cliniques suggérant un phénomène invasif, l’utilisation d’antibactériens à bonne diffusion systémique peut être nécessaire.
La diarrhée chronique n’a pas été suffisamment étudiée avec ce médicament. Le racécadotril n'a pas été évalué au cours des diarrhées associées aux antibiotiques. Par conséquent, le racécadotril ne doit pas être utilisé dans ces cas.
Du fait d'une biodisponibilité potentiellement réduite, le racécadotril ne devra pas être administré en cas de vomissements prolongés ou incontrôlables.
Ce médicament ne doit pas être administré aux nourrissons de moins de 3 mois, car aucune étude clinique n’a été menée dans cette population.
Excipients :
Ce médicament contient 225 mg/dose-kg de saccharose. Ceci est à prendre en compte pour les patients atteints de diabète sucré.
Les patients présentant une intolérance au fructose, un syndrome de malabsorption du glucose et du galactose ou un déficit en sucrase/isomaltase (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Ce médicament contient 0,84 mg de sodium par dose-kg.
La quantité de sodium doit être prise en compte dans l’apport alimentaire quotidien maximum recommandé par l’OMS correspondant à 1500 mg chez les enfants.
Ce médicament contient 1,13 mg de benzoate de sodium par dose-kg.
Le benzoate de sodium peut accroître le risque d’ictère (jaunissement de la peau et des yeux) chez le nouveau-né (jusqu’à 4 semaines).
Ce médicament contient 1,06 mg de propylène glycol par dose-kg.
Insuffisance rénale et hépatique :
En cas d'insuffisance rénale ou hépatique, TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS ne devra pas être administré en raison de l'absence de données.
Hypersensibilités :
Des réactions cutanées ont été rapportées avec l’utilisation de racécadotril. Dans la plupart des cas, ces réactions sont légères et ne requièrent aucun traitement. Cependant dans certaines situations, ces réactions peuvent être sévères et mettre en jeu le pronostic vital. Le lien avec la prise de racécadotril ne peut pas être entièrement exclu. Si des réactions cutanées sévères apparaissent, le traitement par racécadotril doit être immédiatement arrêté.
Des cas d’hypersensibilité et d’œdème de Quincke ont été rapportés chez des patients traités par le racécadotril. Ces évènements peuvent survenir à tout moment au cours du traitement.
Un angioœdème du visage, des extrémités, des lèvres, des muqueuses peut se produire.
Lorsque l’angioœdème est associé à une obstruction des voies respiratoires supérieures, comme par exemple au niveau de la langue, de la glotte et/ou du larynx, un traitement d’urgence doit être immédiatement administré.
Le racécadotril doit être interrompu et le patient doit faire l’objet d’une surveillance médicale étroite avec initiation d’un suivi approprié jusqu’à disparition complète et durable des symptômes. Le racécadotril ne doit pas être réintroduit.
Le racécadotril ou certaines classes thérapeutiques sont susceptibles de provoquer une réaction vasculaire à type d’angioœdème de la face et du cou, résultant de l’inhibition de la dégradation de la bradykinine.
Les conséquences de l’angioœdème peuvent parfois être fatales, par obstruction des voies respiratoires. L’angioœdème peut survenir indépendamment d’une association simultanée entre ces médicaments, au cas où le patient aurait été exposé antérieurement à l’un des deux protagonistes. Il conviendra de rechercher des antécédents de survenue de cet effet et de mesurer la nécessité de ce type d’association.
L’association du racécadotril à certains médicaments majorant la concentration de bradykinine, notamment les inhibiteurs de l’enzyme de conversion (IEC) (p.ex. : perindopril et ramipril) augmente le risque de provoquer un angioœdème bradykinique (voir rubrique 4.5).
Par conséquent, une évaluation rigoureuse du rapport bénéfice / risque est nécessaire avant d'initier le traitement par le racécadotril chez les patients sous inhibiteurs de l'enzyme de conversion (voir rubrique 4.5).
Réactions cutanées indésirables graves (SCAR) :
Des réactions cutanées indésirables graves (SCAR), notamment des syndromes d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) pouvant mettre en jeu le pronostic vital ou être fatales, ont été rapportées avec le traitement par racécadotril. Il faut indiquer aux patients quels sont les signes et symptômes et exercer une surveillance étroite pour déceler d’éventuelles réactions cutanées. En cas d'apparition de signes et de symptômes évocateurs d'un DRESS, il faut immédiatement arrêter le racécadotril et envisager un traitement alternatif. Si un DRESS apparaît chez un(e) patient(e) lors de l’administration de racécadotril, il ne faut en aucun cas reprendre le traitement par racécadotril chez ce/cette patient(e).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Association déconseillée
Médicaments bradykinine et angioœdème
Certains médicaments ou classes thérapeutiques sont susceptibles de provoquer une réaction vasculaire à type angioœdème de la face et du cou, résultant de l’inhibition de la dégradation de la bradykinine. Les médicaments les plus fréquemment impliqués sont les IEC (p.ex. : périndopril, ramipril), et dans une moindre mesure les antagonistes de l’angiotensine II (p.ex. : candésartan, irbésartan), les immunosuppresseurs dits mTORi, des antidiabétiques de la classe des gliptines, le racécadotril, l’estramustine, le sacubitril et l’altépase recombinante.
Les conséquences de l’angioœdème peuvent parfois être fatales, par obstruction des voies respiratoires. L’angioœdème peut survenir indépendamment d’une association simultanée entre ces médicaments, au cas où le patient aurait été exposé antérieurement à l’un des deux protagonistes. Il conviendra de rechercher des antécédents de survenue de cet effet et de mesurer la nécessité de ce type d’association.
Association déconseillée (voir aussi la rubrique 4.4)
+ Autres médicaments à risque d’angioœdème bradykinique (voir la rubrique Médicaments, bradykinine et angioœdème).
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études sur l'animal n'ont montré aucun effet nocif direct ou indirect concernant la toxicité sur la reproduction. Les données cliniques sur l'utilisation du racécadotril au cours de la grossesse sont très limitées. En conséquence TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS ne doit pas être administré au cours de la grossesse quel qu'en soit le terme.
Allaitement
En l'absence de données sur le passage du racécadotril dans le lait et en raison de ses propriétés pharmacologiques et de l'immaturité du tube digestif du nouveau-né, TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS ne doit pas être administré au cours de l'allaitement.
Fertilité
Aucun effet sur la fertilité n'a été observé lors des études de fertilité menées chez les rats mâle et femelle.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS n’a aucun effet ou un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines.
Les essais cliniques conduits sur TIORFAN sachets, une autre forme pharmaceutique pour nourrissons et enfants au cours de la diarrhée aiguë ont fourni des données de sécurité d'emploi chez 860 nourrissons et enfants traités par du racécadotril et 441 traités par du placebo.
Les effets indésirables présentés dans le tableau ci-dessous ont été observés plus fréquemment avec racécadotril qu'avec placebo au cours des essais cliniques ou ont été rapportés pendant la période de commercialisation.
Les effets indésirables sont repris selon les classes principales de systèmes d’organes MedDRA. Au sein de chaque classe de systèmes d’organes, les effets indésirables sont présentés par fréquence. Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité. La fréquence des effets indésirables a été définie selon la convention suivante : très fréquent (≥1/10), fréquent (≥1/100 à <1/10), peu fréquent (≥1/1 000 à <1/100), rare (≥1/10 000 à <1/1 000), très rares (<1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Des réactions cutanées indésirables graves (SCAR), notamment des syndromes d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), ont été rapportées avec le traitement par racécadotril (voir Section 4.4).
|
Classes de systèmes d’organes |
Fréquence |
Effet indésirable |
|
Infections et infestations |
Peu fréquent |
Amygdalite |
|
Affections de la peau et du tissu sous-cutané |
Peu fréquent |
Rash, érythème. |
|
Fréquence indéterminée |
Urticaire, angio-œdème (œdème de Quincke) œdème de la langue, de la face, des lèvres, ou des paupières, érythème polymorphe, érythème noueux, rash papulaire, prurit, prurigo, toxidermie, syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) |
|
|
Affection du système immunitaire |
Fréquence indéterminée |
Choc anaphylactique |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Dans les cas de surdosage rapportés, les patients n'ont pas présenté d'effets indésirables.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : ANTIDIARRHEIQUE ANTISECRETOIRE INTESTINAL.
Code ATC: A07XA04. (A: appareil digestif et métabolisme).
Le racécadotril est une prodrogue qui doit être hydrolysée en son métabolite actif, le thiorphan, qui est un inhibiteur de l'enképhalinase, enzyme de la membrane cellulaire, présente dans différents tissus, dont l'épithélium intestinal.
Cette enzyme contribue à l'hydrolyse de peptides exogènes et endogènes, telles que les enképhalines.
Le racécadotril protège ainsi les enképhalines de la dégradation enzymatique, prolongeant leur action au niveau des synapses enképhalinergiques de l'intestin grêle, réduisant l'hypersécrétion.
Le racécadotril est un antisécrétoire intestinal pur. Il diminue l'hypersécrétion intestinale d'eau et d'électrolytes induite par la toxine cholérique ou l'inflammation, sans avoir d'effet sur la sécrétion basale. Il exerce une activité antidiarrhéique, sans modification de la durée du transit intestinal.
Dans deux études cliniques effectuées chez l'enfant avec les présentations en sachets, le racécadotril réduit de 40% et 46%, respectivement, le poids de selles dans les premières 48 heures.
Une réduction significative de la durée de la diarrhée et du besoin de réhydratation a également été observée.
Une méta-analyse fondée sur les données individuelles (9 essais cliniques randomisés avec les sachets, racécadotril versus placebo, en plus de la solution de réhydratation orale) a recueilli les données individuelles des 1384 garçons et filles souffrant de diarrhée aiguë de sévérité variée, traités en ambulatoire ou en hospitalisation.
L'âge moyen était de 12 mois (intervalle interquartile: 6 à 39 mois).
Un total de 714 patients avait moins de 1 an et 670 patients avaient plus de 1 an. Le poids moyen variait de 7,4 kg à 12,2 kg selon les études. La durée globale moyenne de la diarrhée après l'inclusion était de 2,81 jours dans le groupe placebo et de 1,75 jour pour racécadotril.
Le pourcentage de patients guéris était plus élevé dans les groupes traités par le racécadotril comparativement au placebo [Rapport de risque (HR) : 2,04 ; IC 95% : 1,85 à 2,32 ; p < 0,001; régression des hasards proportionnels de Cox]. Les résultats étaient très semblables pour les nourrissons (< 1 an) (HR : 2,01, IC 95% : 1,71 à 2,36, p < 0,001) et les jeunes enfants (> 1 an) (HR : 2,16, IC 95 % : 1,83 à 2,57, p < 0,001). Pour les essais sur les patients hospitalisés (n = 637), le ratio moyen des selles entre racécadotril et placebo était de 0,59 (IC 95% : 0,51 à 0,74), p < 0,001).
Pour les essais sur patients traités en ambulatoire (n = 695), le ratio moyen de selles avec diarrhée entre racécadotril et placebo était de 0,63 (IC 95% : 0,47 à 0,85, p < 0,001).
Le racécadotril ne provoque pas de ballonnement abdominal. Au cours des essais cliniques, le racécadotril a induit une constipation secondaire selon la même fréquence que le placebo.
Par voie orale, l'activité du racécadotril reste périphérique sans effet sur le système nerveux central.
Une étude clinique randomisée, croisée, a montré que le racécadotril 100 mg à la dose thérapeutique (1 gélule) ou à une dose supérieure (4 gélules) n'induit pas de prolongation du QT/QTc chez 56 adultes volontaires sains (contrairement à l'effet observé avec la moxifloxacine, utilisée comme contrôle positif).
5.2. Propriétés pharmacocinétiques
Absorption
Après administration orale, le racécadotril est rapidement absorbé. L'activité sur l'enképhalinase plasmatique apparaît dès la trentième minute.
La biodisponibilité du racécadotril n'est pas modifiée par les repas, mais le pic d'activité est retardé d'environ 1 heure et demie.
Distribution
Après administration orale de racécadotril marqué au 14C chez des volontaires sains, la concentration de racécadotril était environ 200 fois supérieure dans le plasma que dans les cellules sanguines et environ 3 fois supérieure dans le plasma que dans le volume total de sang. Le racécadotril ne se lie pas aux cellules sanguines de manière importante.
Dans le plasma, le volume apparent moyen de distribution de 66,4 L/kg démontre une distribution modérée du 14C dans les autres tissus.
Quatre-vingt-dix pour cent du métabolite actif du racécadotril, tiorphan, (RS)-N-(1-oxo-2-(mercaptométhyl)-3- phénylpropyl) glycine, sont liés aux protéines plasmatiques, principalement, l'albumine.
Les propriétés pharmacocinétiques du racécadotril ne sont pas modifiées lors de l'administration de doses répétées ou chez le sujet âgé.
L'amplitude et la durée d'action du racécadotril sont liées à la dose administrée. Le pic de concentration est de 2 h 30 environ et correspond à une inhibition de 90 % de l'activité enzymatique pour la dose administrée de 1,5 mg/kg.
Pour une dose de 100 mg, la durée d'activité sur l'enképhalinase plasmatique est d'environ 8 heures.
Métabolisme
La demi-vie biologique du racécadotril, déterminée à partir de l’inhibition plasmatique de l’enképhalinase, est de 3 heures.
Le racécadotril est rapidement hydrolysé en thiorphan (RS)-N-(1-oxo-2-(mercaptométhyl)-3-phénylpropyl) glycine, son métabolite actif, lui-même transformé en métabolites inactifs S-methylthiorphan sulfoxyde, S methyl tiorfan, acide 2-methanesulfinylmethyl propionique et acide 2-methylsulfanylmethyl propionique, qui ont tous été formés à plus de 10% de l'exposition systémique de la molécule mère.
D'autres métabolites mineurs ont également été détectés et quantifiés dans les urines et les matières fécales. L'administration répétée de racécadotril n'induit pas d'accumulation dans l'organisme.
Les données in vitro montrent que, le racécadotril/thiorfan et ses quatre métabolites inactifs majeurs n'agissent pas de manière significative comme inhibiteurs des isoformes des cytochromes CYP 3A4, 2D6, 2C9, 1A2 et 2C19.
Les données in vitro montrent que, le racécadotril/thiorfan et ses quatre métabolites inactifs majeurs n'agissent pas de manière significative comme inducteurs des isoformes du cytochrome CYP (famille 3A, 2A6, 2B6, 2C9/2C19, famille 1A, 2E1) et des enzymes qui se lient à la glucuronyltransférase.
Le racécadotril ne modifie pas la liaison protéique de produits fortement liés aux protéines, tels que tolbutamide, warfarine, acide niflumique, digoxine ou phénytoïne.
Chez des patients insuffisants hépatiques (cirrhose, Child-Pugh B), le profil pharmacocinétique du métabolite montre les mêmes Tmax et T1/2 et de plus faibles Cmax (-65 %) et Aire sous la courbe (-29 %), par rapport à des sujets sains.
Chez des patients ayant une insuffisance rénale sévère (clairance de la créatinine entre 11 et 39 mL/mn), le profil pharmacocinétique du métabolite montre une plus faible Cmax (-49 %) et de plus grandes Aire sous la Courbe (+15 %) et T1/2 par rapport à des volontaires sains (clairance de la créatinine > 70 mL/min).Dans la population pédiatrique, les résultats pharmacodynamiques sont similaires à ceux de la population adulte, avec une Cmax atteinte 2 heures 30 minutes après l'administration. Il n'y a pas d'accumulation après administration de doses répétées toutes les 8 heures, pendant 7 jours.
Excrétion
Le racécadotril est éliminé via ses métabolites actifs et inactifs. L'élimination se fait surtout par voie rénale (81.4%), et à moindre degré par voie fécale (environ 8%). L'excrétion par voie pulmonaire n'est pas significative (moins de 1% de la dose).
5.3. Données de sécurité préclinique
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, génotoxicité ou toxicologie sur les fonctions de reproduction et de développement, n’ont pas révélé de risque particulier pour l’homme.
Des études de toxicité chronique de 4 semaines réalisées chez le singe et le chien, n’ont mis en évidence aucun effet majeur jusqu'à des doses de 250 mg/kg/jour et 200 mg/kg/jour respectivement. L’exposition correspondante chez le singe conduit à une marge de sécurité de 21 et 28 sur la base des ratios AUC et Cmax, respectivement, par rapport à la dose pédiatrique recommandée, soit 4,5 mg/kg/jour. Aux doses plus élevées, les principaux effets indésirables observés chez l’animal étaient une diarrhée abondante, des vomissements, une cétonurie et une anémie sans pertinence clinique connue.
Le racécadotril ne s'est pas révélé immunotoxique chez la souris traitée pendant 1 mois.
Une exposition de plus longue durée (1 an) chez le singe a montré une mortalité significative due à des infections et des réponses réduites en anticorps à la vaccination (à la dose de 500 mg/kg/jour) et une absence d’effets indésirables y compris de type infection et immunosuppression à la dose de 150 mg/kg/jour.
En l’absence de données toxicocinétiques correspondantes, les marges de sécurité basées sur la conversion de la surface corporelle en dose équivalente humaine (DEH) de cette dose sans effet indésirable sont d'au moins 10 par rapport à la dose pédiatrique recommandée.
De même, chez les chiens traités à la dose de 200 mg/kg/jour pendant 26 semaines, quelques infections/réactions immunitaires (nécrose hépatocellulaire d'origine infectieuse, anémie liée à la présence d'anticorps anti-érythrocytaires sans hypoplasie médullaire) et une thrombocytopénie ont été détectées, qui en l’absence de toxicocinétique n’a pas permis le calcul d’une marge de sécurité. Leur signification clinique est inconnue.
Les tests de cancérogénicité n'ont pas été effectués car il s'agit d'un traitement de courte durée.
Une étude de toxicité réalisée chez des rats juvéniles (âgés de 4 à 42 jours) n’a mis en évidence aucun effet significatif dû au racécadotril à des doses jusqu’à 500 mg/kg/jour qui correspond à une marge de sécurité de 63 par rapport à la dose pédiatrique recommandée.
Benzoate de sodium, hydroxyéthylcellulose, gomme xanthane, saccharose, citrate de sodium, acide lactique (pour ajustement du pH), arôme fraise*.
* Composition de l’arôme fraise : composés aromatiques, composés aromatiques d’origine naturelle et propylèneglycol.
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d’autres médicaments.
Avant ouverture du flacon : 24 mois
Après première ouverture du flacon : 10 jours.
6.4. Précautions particulières de conservation
Pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Flacon (PET) de 50 mL muni d’un bouchon sécurité-enfant (PE) avec une seringue de 10 mL pour administration orale graduée en kg. Boîte de 1 flacon contenant 112 doses-kg.
Flacon (PET) de 180 mL muni d’un bouchon sécurité-enfant (PE) avec une seringue de 10 mL pour administration orale graduée en kg. Boîte de 1 flacon contenant 440 doses-kg.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
BIOPROJET PHARMA
9 RUE RAMEAU
75002 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 738 4 8 : Flacon (PET) de 50 mL avec seringue de 10 mL graduée en kg.
· 34009 301 738 5 5 : Flacon (PET) de 180 mL avec seringue de 10 mL graduée en kg.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I
ANSM - Mis à jour le : 17/04/2026
TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable
Racécadotril
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament a été personnellement prescrit pour votre enfant. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si votre enfant ressent un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable ?
3. Comment prendre TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : ANTIDIARRHEIQUE ANTISECRETOIRE INTESTINAL. Code ATC : A07XA04. (A: appareil digestif et métabolisme).
TIORFAN 4 mg/mL suspension buvable est un médicament pour le traitement de la diarrhée.
TIORFAN 4 mg/mL suspension buvable est utilisé en complément de la réhydratation orale et des mesures diététiques dans le traitement des symptômes de la diarrhée aiguë du nourrisson et de l'enfant de plus de 3 mois et pesant 7 kg et plus. Il doit être pris avec autant de liquides qu’ils pourront boire et en association avec les mesures diététiques habituelles lorsque celles-ci ne suffisent pas à elles seules à maîtriser la diarrhée et si on ne peut pas remédier à la cause de la diarrhée.
Lorsque le traitement de la cause est possible, le racécadotril peut être administré en complément de ce traitement.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable?
· Si votre médecin vous a informé(e) d'une intolérance à certains sucres, contactez-le avant de donner ce médicament à votre enfant car il contient du saccharose.
Ne donnez jamais TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS :
· si votre enfant est allergique (hypersensible) à la substance active ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6.
· si une éruption cutanée sévère ou une desquamation de la peau, des cloques et/ou des aphtes sont apparues après que vous avez pris du racécadotril.
Avertissements et précautions
Adressez-vous à votre médecin ou votre pharmacien avant d’administrer TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS à votre enfant si :
· votre enfant a moins de trois mois ou pèse moins de 7 kg,
· vous remarquez la présence de sang ou de pus dans les selles de votre enfant et s’il a de la fièvre. La diarrhée pourrait être causée par une infection bactérienne qui doit être traitée par votre médecin,
· votre enfant a des diarrhées chroniques ou de la diarrhée causée par des antibiotiques,
· votre enfant a plus de 6 selles liquides par jour ou a une diarrhée qui s'accompagne d'une perte de poids,
· votre enfant souffre d’une maladie rénale ou d’un mauvais fonctionnement du foie,
· votre enfant a des vomissements prolongés ou non contrôlés,
· votre enfant souffre du diabète (voir rubrique «TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS contient du sodium, du benzoate de sodium, du saccharose et du propylène glycol »).
Des cas d'hypersensibilité et d’œdème de Quincke (gonflement) ont été rapportés chez des patients traités par le racécadotril (la substance active de ce médicament). Un angioœdème du visage, des extrémités, des lèvres, des muqueuses etc.., ou gonflement des voies respiratoires supérieures, comme la langue, la glotte et/ou le larynx peuvent se produire à tout moment au cours du traitement.
Si vous ressentez un quelconque de ces effets indésirables, arrêtez immédiatement le traitement et contactez votre médecin.
Les patients ayant des antécédents d'angioœdème (gonflement) sans rapport avec le traitement par le racécadotril peuvent présenter un risque accru d'angioœdème.
L’utilisation concomitante de racécadotril et d’autres médicaments peut augmenter le risque d’angioœdème (voir la rubrique « Autres médicaments et TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable»)
Des réactions cutanées ont été rapportées avec l’utilisation de racécadotril. Dans la plupart des cas, ces réactions sont légères et modérées. Si des réactions cutanées sévères apparaissent, le traitement par racécadotril doit être immédiatement arrêté. Le racécadotril ne doit pas être réintroduit.
Ce traitement est administré en complément de la réhydratation orale et des mesures diététiques. Votre médecin décidera si votre enfant a besoin d'une solution de réhydratation orale. Vous devrez alors suivre les conditions d'utilisation de la solution de réhydratation orale prescrite par votre médecin et suivre les conseils concernant l’alimentation.
Des réactions cutanées graves, notamment des syndromes d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), ont été rapportées avec le traitement par racécadotril. Arrêtez d'utiliser le racécadotril et consultez immédiatement un médecin si vous remarquez l'un des symptômes liés à ces réactions cutanées graves décrites à la Section 4.
Autres médicaments et TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS, suspension buvable
Informez votre médecin ou votre pharmacien si votre enfant prend, a pris récemment ou pourrait prendre tout autre médicament, notamment :
· un inhibiteur de l’enzyme de conversion (par exemple : périndopril ou ramipril) pour faire baisser la pression artérielle et faciliter le travail du cœur,
· les antagonistes de l’angiotensine II (par exemple : candésartan ou irbésartan) pour traiter l’hypertension artérielle et l’insuffisance cardiaque.
Si vous donnez ou avez donné récemment un autre médicament à votre enfant, y compris un médicament sans ordonnance, parlez-en à votre médecin ou à votre pharmacien.
Grossesse, allaitement et fertilité
Grossesse
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Compte tenu des données disponibles, ce médicament n’est pas recommandé au cours de la grossesse quel qu'en soit le terme.
Allaitement
En l'absence de données sur le passage de la substance active dans le lait maternel ce médicament ne doit pas être administré au cours de l'allaitement.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Ce médicament n’a aucun effet ou un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines.
TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS contient du sodium, du benzoate de sodium, du saccharose et du propylène glycol
Si votre médecin vous a informé que votre enfant présente une intolérance à certains sucres, demandez-lui conseil avant de donner Tiorfan 4 mg/mL Nourrissons et Enfants à votre enfant.
Ce médicament contient 225 mg de saccharose par dose-kg. Ceci est à prendre en compte pour les patients atteints de diabète sucré.
Les patients ayant une intolérance au fructose, un syndrome de malabsorption du glucose et du galactose ou un déficit en sucrase/isomaltase (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Ce médicament contient 0,84 mg de sodium (composant principal du sel de cuisine/table) par dose-kg.
La quantité de sodium doit être prise en compte dans l’apport alimentaire quotidien maximum recommandé par l’OMS correspondant à 1500 mg chez les enfants.
Ce médicament contient 1,13 mg de benzoate de sodium par dose-kg.
Le benzoate de sodium peut accroître le risque d’ictère (jaunissement de la peau et des yeux) chez le nouveau-né (jusqu’à 4 semaines).
Ce médicament contient 1,06 mg de propylène glycol par dose-kg.
3. COMMENT PRENDRE TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS suspension buvable?
Veillez à toujours donner ce médicament à votre enfant en suivant exactement les instructions de cette notice ou les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Ce médicament se présente sous forme d’une suspension buvable avec une odeur caractéristique de fraise.
Réservé aux nourrissons et aux enfants de 7 kg à 52 kg.
La dose recommandée est établie en fonction du poids corporel de votre enfant. Elle est de 1,5 mg/kg/prise (ce qui correspond à 1 dose-kg).
Le premier jour : donner la première prise immédiatement à votre enfant puis selon l’heure de la première prise, donner jusqu’à un maximum de 2 prises supplémentaires réparties dans la journée, sans dépasser 3 prises dans la journée. Les prises doivent se faire de préférence au début des trois principaux repas.
Les jours suivants : donner 3 prises réparties dans la journée, de préférence au début des trois principaux repas.
La posologie journalière maximale totale est de 3 prises.
Le médicament s’administre au moyen de la seringue pour administration orale (graduée en kg de poids corporel) qui délivre une dose 1,5 mg de racécadotril par graduation indiquée en kg.
Pour chaque prise :
· Nourrissons et enfants jusqu’à 26 kg : utiliser la seringue en remplissant jusqu’à la graduation indiquant le poids du nourrisson ou de l’enfant.
· Enfants entre 27 kg et 38 kg (voir tableau ci-dessous) : remplir une première fois la seringue jusqu’à la graduation 13 kg et donner la suspension à votre enfant. Puis remplir une deuxième fois la seringue jusqu’à atteindre un total égal au poids de l’enfant et donner à nouveau la suspension à votre enfant.
· Enfants entre 39 kg et 52 kg (voir tableau ci-dessous) : remplir une première fois la seringue de 10 mL jusqu’à la graduation 26 kg et donner la suspension à votre enfant. Puis remplir une deuxième fois la seringue de 10 mL jusqu’à atteindre un total égal au poids de l’enfant et donner à nouveau la suspension à votre enfant.
· Au-delà de 52 kg, il convient d’utiliser des formes pharmaceutiques plus adaptées.
|
Poids de l’enfant |
Graduation pour le premier remplissage de la seringue |
Graduation pour le 2ème remplissage de la seringue |
|
27 kg |
13 kg |
14 kg |
|
28 kg |
13 kg |
15 kg |
|
29 kg |
13 kg |
16 kg |
|
30 kg |
13 kg |
17 kg |
|
31 kg |
13 kg |
18 kg |
|
32 kg |
13 kg |
19 kg |
|
33 kg |
13 kg |
20 kg |
|
34 kg |
13 kg |
21 kg |
|
35 kg |
13 kg |
22 kg |
|
36 kg |
13 kg |
23 kg |
|
37 kg |
13 kg |
24 kg |
|
38 kg |
13 kg |
25 kg |
|
39 kg |
26 kg |
13 kg |
|
40 kg |
26 kg |
14 kg |
|
41 kg |
26 kg |
15 kg |
|
42 kg |
26 kg |
16 kg |
|
43 kg |
26 kg |
17 kg |
|
44 kg |
26 kg |
18 kg |
|
45 kg |
26 kg |
19 kg |
|
46 kg |
26 kg |
20 kg |
|
47 kg |
26 kg |
21 kg |
|
48 kg |
26 kg |
22 kg |
|
49 kg |
26 kg |
23 kg |
|
50 kg |
26 kg |
24 kg |
|
51 kg |
26 kg |
25 kg |
|
52 kg |
26 kg |
26 kg |
Durée du traitement
Votre médecin vous indiquera la durée du traitement par TIORFAN 4 mg/mL, suspension buvable. Le traitement sera poursuivi jusqu'au retour de deux selles moulées consécutives, sans dépasser 7 jours.
Mode d’administration
Voie orale.
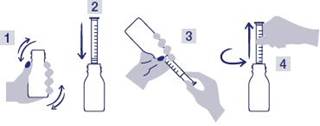
1) Agiter vigoureusement le flacon avant l’emploi. Schéma 1
2) Ouvrir le flacon en tournant et en appuyant sur le bouchon sécurité-enfant
3) Introduire à fond la seringue dans l’embout de prélèvement. Schéma 2
4) Pour remplir la seringue, tenir le flacon « tête en bas ». Bien maintenir la seringue en place et tirer doucement et régulièrement le piston jusqu’à la graduation nécessaire en kg. Schéma 3
5) Remettre le flacon « tête en haut » et retirer la seringue. Schéma 4
6) Maintenir l’enfant en position debout pendant l’administration. Introduire la seringue dans la bouche de l’enfant sans l’enfoncer, en la dirigeant sur la face interne de la joue. Administrer la totalité de la suspension en appuyant doucement et progressivement sur le piston.
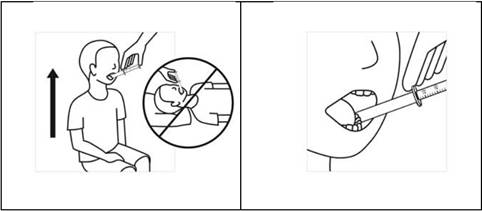
7) Après chaque utilisation, démonter la seringue pour administration orale, la rincer à l’eau et la sécher.
L’usage de cette seringue pour administration orale est strictement réservé à l’administration de TIORFAN 4 mg/mL dose-kg.
Pour compenser la déshydratation due à la diarrhée, vous devez prendre ce médicament avec une quantité suffisante de liquide et de sels (électrolytes). Pour remplacer au mieux la perte de liquide et de sels, il est recommandé d’utiliser une solution de réhydratation orale (En cas de doute, consulter votre médecin ou votre ou votre pharmacien).
Si vous avez donné plus de TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS que vous n’auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez de donner TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS :
Ne donnez pas de dose double pour compenser la dose que vous avez oublié de donner à votre enfant. Poursuivez avec la dose suivante.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Vous devez arrêter de donner TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS à votre enfant et consulter immédiatement un médecin si votre enfant ressent des symptômes d’angio-œdème tels que :
o Gonflement du visage, de la langue ou du pharynx
o Difficulté pour avaler
o Urticaires et difficultés pour respirer.
Arrêtez d'utiliser le racécadotril et consultez immédiatement un médecin si vous remarquez l'un des symptômes suivants :
· Éruption cutanée généralisée, température corporelle élevée et ganglions lymphatiques hypertrophiés (syndrome DRESS)
· Difficulté à respirer, gonflement, étourdissements/vertiges, rythme cardiaque rapide, transpiration et sensation de perte de conscience ; ce sont les symptômes d'une réaction allergique soudaine et grave
Effets indésirables peu fréquents (rapportés chez au moins 1 patient sur 1 000 mais chez moins de 1 patient sur 100) :
Amygdalite (Inflammation des amygdales), rash (éruptions cutanées) et érythème (rougeur de la peau)
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
Erythème polymorphe (lésions rosâtres au niveau des extrémités et dans la bouche), œdème de la langue, œdème des lèvres, œdème des paupières, œdème du visage, angio-œdème (inflammation souscutanée touchant diverses parties du corps), urticaire, érythème noueux (inflammation prenant la forme d'un nodule sous la peau), rash papulaire (éruption cutanée présentant des petites lésions dures et pustuleuses), prurit (démangeaisons affectant tout le corps), prurigo (lésions cutanées provoquant des démangeaisons).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS suspension buvable?
Tenir ce médicament hors de la vue et de la portée des enfants.
Ne donnez pas ce médicament après la date de péremption indiquée sur le flacon. La date de péremption fait référence au dernier jour de ce mois.
N’utilisez pas ce médicament si vous remarquez des signes visibles de détérioration.
Une fois votre traitement terminé, rapportez à votre pharmacien la boîte y compris la seringue pour administration orale ainsi que le flacon pour une destruction correcte et appropriée de ce médicament.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient TIORFAN 4 mg/mL NOURRISSONS ET ENFANTS suspension buvable
· La substance active est :
Racécadotril............................................................................................................................ 4 mg
Pour 1 mL de suspension buvable
· Les autres composants sont :
Benzoate de sodium, hydroxyéthylcellulose, gomme xanthane, saccharose, citrate de sodium, acide lactique (pour ajustement du pH), arôme fraise (contenant notamment du propylène glycol). Voir rubrique 2.
Ce médicament se présente sous forme d’une suspension buvable avec une odeur caractéristique de fraise.
Conditionnements :
Flacon PET de 50 mL avec bouchon sécurité-enfant et seringue de 10 mL graduée en kg. Boîte de 1.
Flacon PET de 180 mL avec bouchon sécurité-enfant et seringue de 10 mL graduée en kg Boîte de 1.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
BIOPROJET PHARMA
9 RUE RAMEAU
75002 PARIS
Exploitant de l’autorisation de mise sur le marché
BIOPROJET PHARMA
9 RUE RAMEAU
75002 PARIS
UNITHER LIQUID MANUFACTURING
1-3 ALLEE DE LA NESTE
31770 COLOMIERS
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[À compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).