Dernière mise à jour le 03/08/2026
COPAXONE 20 mg/ml, solution injectable en seringue préremplie
Indications thérapeutiques
COPAXONE est un médicament indiqué dans le traitement des formes rémittentes de sclérose en plaques (SEP). Il modifie la manière dont fonctionne le système immunitaire de votre corps et il est classé comme agent immunomodulateur. On attribue les symptômes de la SEP à un dysfonctionnement du système immunitaire de l’organisme. Il en résulte une inflammation sous forme de plaques au niveau du cerveau et de la moelle épinière.
COPAXONE est utilisé pour réduire le nombre de poussées lors de la SEP (aggravations). Son efficacité n’a pas été démontrée si vous présentez une forme de SEP autre que rémittente et si vous ne présentez que peu ou pas de poussée. COPAXONE peut n’avoir aucun effet sur la durée ou sur la sévérité d’une poussée.
Il est indiqué pour le traitement de patients capables de marcher sans aide.
COPAXONE peut également être utilisé chez des patients ayant présenté pour la première fois, des symptômes évocateurs d'un risque élevé de développer une SEP. Avant de débuter le traitement, votre médecin devra éliminer toute autre cause médicale pouvant expliquer ces symptômes.
Présentations
> 28 seringue(s) préremplie(s) en verre de 1 ml
Code CIP : 34009 363 840 1 9
Déclaration de commercialisation : 03/07/2017
Cette présentation n'est pas agréée aux collectivités
- Prix hors honoraire de dispensation : 542,40 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 543,42 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
Autres informations
- Titulaire de l'autorisation : TEVA SANTE
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription initiale réservée à certains spécialistes
- renouvellement de la prescription réservé aux spécialistes en NEUROLOGIE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 008 206 2
ANSM - Mis à jour le : 17/11/2025
COPAXONE 20 mg/ml, solution injectable en seringue préremplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Glatiramère............................................................................................................................ 18 mg
sous forme d’acétate de glatiramère*...................................................................................... 20 mg
Pour 1 seringue préremplie (1 ml).
* L'acétate de glatiramère est le sel acétate de polypeptides synthétiques comprenant 4 acides aminés naturels : acide L-glutamique, L-alanine, L-tyrosine et L-lysine avec une fraction molaire moyenne de respectivement 0,129-0,153, 0,392-0,462, 0,086-0,100 et 0,300-0,374. Le poids moléculaire moyen de l'acétate de glatiramère est compris entre 5 000 et 9 000 daltons. En raison de la complexité de sa composition, aucun polypeptide spécifié ne peut être totalement caractérisé, y compris en termes de séquence d'acides aminés bien que la composition finale de l'acétate de glatiramère ne soit pas entièrement aléatoire.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution limpide, exempte de particules visibles.
La solution pour injection a un pH de 5,5-7,0 et une osmolarité de 265 mOsmol/L.
4.1. Indications thérapeutiques
COPAXONE n'est pas indiqué dans le traitement des formes progressives d'emblée ou secondairement progressives de sclérose en plaques.
4.2. Posologie et mode d'administration
Posologie
La posologie recommandée chez l'adulte est de 20 mg d'acétate de glatiramère (une seringue préremplie de COPAXONE 20 mg/ml) administrés par voie sous-cutanée une fois par jour.
En l'état actuel des connaissances, la durée de traitement ne peut être précisée.
La décision d'un traitement de longue durée sera prise sur la base d'une évaluation clinique personnalisée au cas par cas par le médecin traitant (neurologue ou médecin expérimenté dans le traitement de la SEP).
Insuffisants rénaux
COPAXONE n'a pas été étudié chez l'insuffisant rénal (voir rubrique 4.4).
Personnes âgées
COPAXONE n'a pas été étudié chez les personnes âgées.
Population pédiatrique
La sécurité et l’efficacité de l’acétate de glatiramère chez les enfants et les adolescents n’ont pas été établies.
Cependant, des données publiées limitées suggèrent que le profil de sécurité chez l'adolescent âgé de 12 à 18 ans traité par 20 mg de COPAXONE par voie sous-cutanée tous les jours est comparable à celui observé chez l'adulte. L’information disponible sur l’utilisation de COPAXONE chez les enfants de moins de 12 ans n’est pas suffisante pour recommander son utilisation. COPAXONE ne doit donc pas être utilisé dans cette population.
Mode d’administration
COPAXONE doit être administré par voie sous-cutanée.
Les patients doivent être formés à la technique d'auto-injection. Ils doivent être surveillés par un professionnel de la santé lors de leur première auto-injection et pendant les 30 minutes qui suivent.
Un site différent doit être choisi pour chaque injection, ce qui réduira les risques d'irritation ou de douleur au site d'injection. Les sites pour l’auto-injection comprennent l'abdomen, les bras, les hanches et les cuisses.
Le dispositif CSYNC est à disposition des patients souhaitant effectuer leur injection à l’aide d’un dispositif d’injection. Le dispositif CSYNC est un auto-injecteur qui doit être utilisé avec les seringues pré-remplies de COPAXONE et il n’a pas été testé avec d’autres seringues pré-remplies. Le dispositif CSYNC doit être utilisé selon le mode d’emploi fourni par le fabricant du dispositif.
Copaxone est contre-indiqué dans les cas suivants :
· Hypersensibilité à la substance active (acétate de glatiramère) ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
COPAXONE doit être administré uniquement par voie sous-cutanée. Les voies intraveineuse ou intramusculaire ne doivent pas être utilisées.
L’acétate de glatiramère peut provoquer des réactions post-injection ainsi que des réactions anaphylactiques (voir rubrique 4.8) :
Réactions post-injection
Le médecin doit expliquer au patient que dans les minutes suivant l'injection de COPAXONE une réaction peut survenir, associant un ou plusieurs des symptômes suivants : vasodilatation (bouffée vasomotrice), douleur thoracique, dyspnée, palpitations ou tachycardie (voir rubrique 4.8). La majorité de ces symptômes est généralement transitoire et disparait spontanément sans séquelle. Si un effet indésirable sévère survient, le patient doit immédiatement arrêter le traitement par COPAXONE et contacter son médecin ou un service médical d’urgence. Un traitement symptomatique adapté pourra être instauré si nécessaire.
Il n’y a pas de données suggérant qu’une population particulière de patients est plus à risque de présenter de telles réactions. Cependant, la prudence est requise lorsque l'on administre COPAXONE à des patients ayant des antécédents d’affections cardiaques. Ces patients doivent être suivis régulièrement durant le traitement.
Réactions anaphylactiques
Des réactions anaphylactiques peuvent se produire peu de temps après l’administration d’acétate de glatiramère, mais également plusieurs mois voire plusieurs années après l’instauration du traitement (voir rubrique 4.8). Des cas d’issue fatale ont été rapportés. Certains signes et symptômes peuvent être communs aux réactions anaphylactiques et aux réactions post-injection.
Tous les patients traités par COPAXONE et leurs aidants doivent être informés des signes et symptômes spécifiques des réactions anaphylactiques et de la nécessité de recevoir des soins médicaux d’urgence en cas de survenue de ces symptômes (voir rubrique 4.8).
En cas de réaction anaphylactique, le traitement par COPAXONE doit être arrêté (voir rubrique 4.3).
Des anticorps anti-acétate de glatiramère ont été détectés dans le sérum de patients traités au long cours par COPAXONE. Les taux maximaux ont été atteints en moyenne après 3 à 4 mois de traitement, puis ces taux ont diminué et se sont stabilisés à un niveau légèrement supérieur à la valeur initiale.
Il n’y a pas de données disponibles suggérant que ces anticorps anti-acétate de glatiramère soient de type neutralisant ou que leur production puisse altérer l'efficacité clinique de COPAXONE.
Chez les patients insuffisants rénaux, la fonction rénale doit être surveillée tant qu'ils sont traités par COPAXONE. Bien que l'existence de dépôt glomérulaire de complexes immuns n'ait pas été démontrée, cette possibilité ne peut être exclue.
De rares cas de lésion hépatique sévère ont été observés (y compris hépatite avec ictère, insuffisance hépatique et, dans des cas isolés, transplantation du foie). Une lésion hépatique est survenue de quelques jours à quelques années après le début du traitement par COPAXONE. Dans la plupart des cas, les lésions hépatiques sévères se sont résolues avec l’arrêt du traitement. Dans certains cas, ces réactions se sont produites dans un contexte de consommation excessive d’alcool, une lésion hépatique existante ou des antécédents de lésion hépatique et l’utilisation d’autres médicaments potentiellement hépatotoxiques. Les patients doivent faire l’objet d’une surveillance régulière afin de déceler tout signe de lésion hépatique et ils doivent recevoir pour instruction de consulter immédiatement un médecin en cas de symptômes de lésion hépatique. En cas de lésion hépatique cliniquement significative, l’arrêt du traitement par COPAXONE doit être envisagé.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les interactions entre COPAXONE et d'autres médicaments n'ont pas été évaluées de façon systématique.
Les observations issues des études cliniques et de l’expérience acquise depuis la commercialisation ne suggèrent pas d’interaction significative entre COPAXONE et les traitements couramment utilisés chez les patients atteints de SEP, y compris lors de l’utilisation concomitante de corticostéroïdes pendant une durée allant jusqu’à 28 jours.
Des études in vitro suggèrent que l'acétate de glatiramère est fortement lié aux protéines plasmatiques, mais n'est pas déplacé par et ne déplace pas la phénytoïne ou la carbamazépine. Néanmoins, dans la mesure où COPAXONE possède théoriquement la capacité d'affecter la distribution des substances se liant aux protéines plasmatiques, l’utilisation concomitante de tels médicaments doit être étroitement surveillée.
4.6. Fertilité, grossesse et allaitement
Grossesse
Un grand nombre de données sur les femmes enceintes (plus de 1 000 issues de grossesses exposées) indiquent une absence de toxicité malformative ou fœtale/néonatale.
COPAXONE peut être utilisé pendant la grossesse, si cela est cliniquement nécessaire.
Les propriétés physico-chimiques et la faible absorption orale suggèrent que l’exposition des nouveau-nés/nourrissons à l’acétate de glatiramère par le lait maternel humain est négligeable. Une étude rétrospective non interventionnelle menée auprès de 60 nourrissons allaités de mères exposées à l’acétate de glatiramère, comparés à 60 nourrissons allaités de mères non exposées à un traitement modificateur de la maladie, et des données limitées recueillies après la commercialisation chez l’être humain, n’ont révélé aucun effet négatif de l’acétate de glatiramère.
COPAXONE peut être utilisé pendant l’allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Aucune étude sur les effets sur l’aptitude à conduire et à utiliser des machines n’a été réalisée.
Dans toutes les études cliniques, les réactions aux sites d'injection ont été les effets indésirables les plus fréquents et ont été rapportées par une majorité de patients traités par COPAXONE. Dans les études contrôlées, le pourcentage de patients ayant rapporté ces réactions au moins une fois, était plus important dans les groupes traités par COPAXONE par rapport au groupe placebo (70 % versus 37 %). Les réactions aux sites d'injection les plus fréquemment rapportées dans les études cliniques et depuis la commercialisation étaient : érythème, douleur, induration, prurit, œdème, inflammation, hypersensibilité et de rares cas de lipoatrophie et de nécrose cutanée.
Des réactions immédiates post-injection ont été décrites. Elles comprenaient au moins un ou plusieurs des symptômes suivants : vasodilatation (bouffée vasomotrice), douleur thoracique, dyspnée, palpitation ou tachycardie (voir rubrique 4.4). Une telle réaction peut survenir dans les minutes qui suivent l'injection de COPAXONE. Au moins un symptôme caractérisant cette réaction immédiate post-injection a été rapporté au moins une fois par 31 % des patients recevant COPAXONE comparé à 13 % dans le groupe placebo.
Les effets indésirables identifiés dans les essais cliniques et l’expérience post-marketing sont présentés dans le tableau ci-après. Les données issues des essais cliniques sont issus de 4 essais pivots en double aveugle contre placebo, au cours desquels 512 patients ont reçu COPAXONE et 509 ont reçu un placebo pendant une durée allant jusqu'à 36 mois. Trois essais portant sur la SEP (sclérose en plaques) de type récurrente/rémittente (SEP-RR) ont inclus un total de 269 patients traités par COPAXONE et 271 patients ayant reçu le placebo, pendant une durée allant jusqu'à 35 mois. Le quatrième essai, réalisé chez des patients qui avaient présenté un premier évènement clinique et qui étaient considérés comme présentant un risque élevé de développer une SEP cliniquement définie, incluait 243 patients traités par COPAXONE et 238 patients ayant reçu le placebo pendant une durée allant jusqu'à 36 mois.
|
Classe de systèmes d'organes (SOC) |
Très fréquent (≥ 1/10) |
Fréquent (≥ 1/100, < 1/10) |
Peu fréquent (≥ 1/1 000, < 1/100) |
Rare (≥ 1/10 000, < 1 000) |
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) |
|
Infections et infestations |
Infection, grippe |
Bronchite, gastro-entérite, infection à Herpès Simplex, otite moyenne, rhinite, abcès dentaire, candidose vaginale* |
Abcès, cellulite, furoncle, zona, pyélonéphrite |
|
|
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
|
Tumeur cutanée bénigne, tumeur |
Cancer cutané. |
|
|
|
Affections hématologiques et du système lymphatique |
|
Lymphadénopathie* |
Leucocytose, leucopénie, splénomégalie, thrombocytopénie, anomalies morphologiques des lymphocytes |
|
|
|
Affections du système immunitaire |
|
Hypersensibilité |
Réaction anaphylactique |
|
|
|
Affections endocriniennes |
|
|
Goitre, hyperthyroïdie |
|
|
|
Troubles du métabolisme et de la nutrition |
|
Anorexie, prise de poids* |
Intolérance à l'alcool, goutte, hyperlipidémie, augmentation du sodium sanguin, diminution de la ferritine sérique |
|
|
|
Affections psychiatriques |
Anxiété*, dépression |
Nervosité |
Rêves anormaux, état confusionnel, état euphorique, hallucination, hostilité, manie, trouble de la personnalité, tentative de suicide |
|
|
|
Affections du système nerveux |
Céphalées |
Dysgueusie, hypertonie, migraine, trouble de l'élocution, syncope, tremblements* |
Syndrome du canal carpien, trouble cognitif, convulsions, dysgraphie, dyslexie, dystonie, trouble moteur, myoclonie, névrite, blocage neuromusculaire, nystagmus, paralysie, paralysie du nerf sciatique, stupeur, déficit du champ visuel |
|
|
|
Affections oculaires |
|
Diplopie, affection oculaire* |
Cataracte, lésion de la cornée, sécheresse oculaire, hémorragie oculaire, ptosis, mydriase, atrophie optique |
|
|
|
Affections de l’oreille et du labyrinthe |
|
Affection de l’oreille |
|
|
|
|
Affections cardiaques |
|
Palpitations*, tachycardie* |
Extrasystoles, bradycardie sinusale, tachycardie paroxystique |
|
|
|
Affections vasculaires |
Vasodilatation* |
|
Varice |
|
|
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée* |
Toux, rhinite saisonnière |
Apnée, épistaxis, hyperventilation, laryngospasme, affection pulmonaire, sensation d’étouffement |
|
|
|
Affections gastro-intestinales |
Nausées* |
Affection ano-rectale, constipation, caries dentaires, dyspepsie, dysphagie, incontinence fécale, vomissements* |
Colite, polype du colon, entérocolite, éructation, ulcère œsophagien, parodontite, hémorragie rectale, augmentation du volume des glandes salivaires |
|
|
|
Affections hépatobiliaires |
|
Anomalies des tests hépatiques |
Cholélithiase, hépatomégalie |
Hépatite toxique, lésion hépatique |
Insuffisance hépatique# |
|
Affections de la peau et du tissu sous-cutané |
Éruption cutanée* |
Ecchymose, hyperhidrose, prurit, affection cutanée*, urticaire |
Angioedème, dermite de contact, érythème noueux, nodule cutané |
|
|
|
Affections musculo-squelettiques et systémiques |
Arthralgie, dorsalgie* |
Cervicalgie |
Arthrite, bursite, douleur du flanc, atrophie musculaire, ostéoarthrite |
|
|
|
Affections du rein et des voies urinaires |
|
Impériosité mictionnelle, pollakiurie, rétention urinaire |
Hématurie, néphrolithiase, affection du tractus urinaire, anomalie des urines |
|
|
|
Affections des organes de reproduction et du sein |
|
|
Engorgement mammaire, dysfonctionnement érectile, prolapsus pelvien, priapisme, affections prostatiques, frottis cervical anormal, affections testiculaires, hémorragie vaginale, affection vulvo-vaginale |
|
|
|
Troubles généraux et anomalies au site d'administration |
Asthénie, douleur thoracique*, réactions au site d'injection*§, douleur* |
Frissons*, œdème de la face*, atrophie au site d'injection♣, réaction locale*, œdème périphérique, œdème, fièvre |
Kyste, sensation de « gueule de bois », hypothermie, réaction immédiate post-injection, inflammation, nécrose au site d'injection, affection des muqueuses |
|
|
|
Lésions, intoxications et complications liées aux procédures |
|
|
Syndrome post-vaccinal |
|
|
* Incidence supérieure de plus de 2 % (> 2/100) dans le groupe traité par COPAXONE par rapport au groupe placebo. La différence d’incidence des effets indésirables sans le symbole * est inférieure ou égale à 2 % entre le groupe traité par COPAXONE et le groupe placebo.
§ Le terme « réactions au site d'injection » (différents types) reprend tous les effets indésirables survenant au site d'injection, à l'exception de l'atrophie au site d'injection et de la nécrose au site d'injection, qui sont présentées séparément dans le tableau.
♣ Comprend des termes qui correspondent à une lipoatrophie localisée aux sites d'injection.
# Quelques cas de transplantation hépatique ont été rapportés.
Dans le quatrième essai mentionné ci-dessus, une phase de traitement en ouvert a suivi la phase contrôlée (voir rubrique 5.1). Aucune modification du profil de sécurité connu de COPAXONE n'a été observée pendant la période de suivi en ouvert allant jusqu'à 5 ans.
Description de certains effets indésirables
Des réactions anaphylactiques peuvent se produire peu de temps après l’administration d’acétate de glatiramère, mais également plusieurs mois voire plusieurs années après l’instauration du traitement (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Symptômes
Quelques cas de surdosage avec COPAXONE (jusqu'à 300 mg d'acétate de glatiramère) ont été rapportés. Ces cas n’ont été associés à aucun effet indésirable autre que ceux mentionnés dans la rubrique 4.8.
Prise en charge
En cas de surdosage, le patient doit être suivi et un traitement symptomatique et de soutien approprié doit être instauré.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le mécanisme d'action par lequel l'acétate de glatiramère exerce ses effets thérapeutiques dans les formes rémittentes de SEP n'est pas entièrement élucidé mais l’on suppose que l’acétate de glatiramère impliquerait une modulation du système immunitaire.
Des études menées chez l’animal et chez des patients atteints de SEP suggèrent que l’acétate de glatiramère agit sur les cellules responsables de l’immunité innée, notamment les monocytes, les cellules dendritiques et les lymphocytes B, qui modulent les fonctions adaptatives des lymphocytes B et T induisant une sécrétion anti-inflammatoire et régulatrice des cytokines. On ne sait pas si l'effet thérapeutique est médié par les effets cellulaires décrits ci-dessus car la physiopathologie de la SEP n'est que partiellement comprise.
Efficacité et sécurité clinique
· SEP-RR :
Un total de 269 patients ont été traités par COPAXONE dans trois études cliniques contrôlées. La première était une étude d'une durée de deux ans chez 50 patients (COPAXONE n = 25, placebo n = 25) avec un diagnostic de SEP de type récurrente/rémittente, établi selon les critères standards de l'époque et qui avaient donc eu au moins deux poussées récurrentes de troubles neurologiques (aggravations) au cours des deux années précédentes. Dans la seconde étude, les critères d'inclusion étaient identiques et 251 patients ont été inclus et traités pendant une durée allant jusqu'à 35 mois (COPAXONE n = 125, placebo n = 126). La troisième étude d’une durée de 9 mois, a porté sur 239 patients (COPAXONE n = 119, placebo n = 120) et les critères d'inclusion étaient similaires à ceux des deux premières études avec un critère additionnel : les patients devaient avoir au moins une lésion rehaussée par le gadolinium sur l'IRM de sélection.
Dans les études cliniques chez les patients atteints de SEP traités par COPAXONE, il y a eu une réduction importante du nombre de poussées par rapport au placebo.
Dans l'étude contrôlée la plus large, le taux de poussées a été réduit de 32 %, de 1,98 sous placebo à 1,34 sous acétate de glatiramère.
Les données d'exposition sont disponibles jusqu'à douze ans chez 103 patients traités par COPAXONE.
COPAXONE a également montré des effets bénéfiques versus placebo sur des paramètres d’IRM pertinents dans la SEP de type récurrente/rémittente.
COPAXONE 20 mg/ml : au cours de l'étude contrôlée 9001/9001E, ayant recruté 251 patients, suivis jusqu’à 35 mois (incluant l'étude 9001E qui est la phase d’extension en aveugle de l’étude 9001), le pourcentage cumulé de patients ayant une progression confirmée du handicap à 3 mois était de 29,4 % dans le groupe placebo versus 23,2 % dans le groupe COPAXONE (p = 0,199).
Il n'a pas été démontré d’effet bénéfique de COPAXONE sur la durée ou la sévérité des poussées.
Actuellement, il n’existe pas de données démontrant l'intérêt de COPAXONE chez les patients atteints de sclérose en plaques de formes primaire progressive ou secondairement progressive.
· Événement clinique unique évocateur d'une SEP
Un essai clinique contrôlé versus placebo a été réalisé chez 481 patients (COPAXONE n = 243, placebo n = 238) ayant présenté un événement clinique bien défini, isolé avec une manifestation neurologique monofocale et des résultats d'IRM hautement évocateurs d'une SEP (au moins deux lésions cérébrales au-dessus de 6 mm de diamètre sur l'IRM en T2). Toute maladie autre que la SEP expliquant mieux la symptomatologie du patient devait être exclue. La phase contrôlée versus placebo a été suivie par une phase de traitement en ouvert. Les patients ayant présenté des symptômes de SEP ou ayant été asymptomatiques pendant trois ans, quel que soit l’événement survenu en premier, ont reçu le traitement médicamenteux actif dans la phase d’extension en ouvert pendant une période supplémentaire de deux ans, sans excéder une durée totale maximale de traitement de 5 ans. Sur les 243 patients initialement randomisés dans le groupe COPAXONE, 198 ont poursuivi le traitement par COPAXONE dans la phase en ouvert. Sur les 238 patients initialement randomisés dans le groupe placebo, 211 ont ensuite été traités par COPAXONE dans la phase en ouvert.
Lors de la phase contrôlée versus placebo sur une période de 3 ans, COPAXONE a permis de retarder, de façon statistiquement significative et cliniquement confirmée, l'évolution vers une sclérose en plaques cliniquement définie (SEPCD) en accord avec les critères de Poser, ceci correspondant à une réduction du risque de 45 % (risque estimé = 0,55, IC à 95 % [0,40-0,77], p = 0,0005). Le taux de patients ayant évolué vers une SEPCD était de 43 % dans le groupe placebo versus 25 % dans le groupe COPAXONE.
L’effet bénéfique de COPAXONE versus placebo a aussi été démontré sur deux critères secondaires à l’IRM : le nombre de nouvelles lésions T2 et le volume des lésions T2.
Une analyse post-hoc a été faite chez des patients ayant des caractéristiques différentes à l’inclusion pour identifier une population présentant un risque élevé de développer une seconde poussée de SEP. Chez les sujets présentant à l'IRM au moins une lésion en T1 prenant le gadolinium et 9 lésions ou plus en T2, 50 % des patients du groupe placebo versus 28 % dans le groupe COPAXONE ont évolué vers une SEPCD au bout de 2,4 ans. Chez les sujets présentant 9 lésions ou plus en T2 à l’inclusion, le taux de conversion en une SEPCD était de 45 % dans le groupe placebo versus 26 % dans le groupe COPAXONE au bout de 2,4 ans. Cependant, l'impact d'un traitement précoce avec COPAXONE sur l'évolution à long terme de la maladie n'est pas connu même dans ces sous-groupes présentant un risque élevé, car l'étude était conçue principalement pour évaluer le délai d'apparition d'un second évènement clinique. Dans tous les cas, le traitement ne devrait être envisagé que pour les patients classés à haut risque.
L'effet démontré dans la phase contrôlée versus placebo s'est maintenu dans la période de suivi à long terme, allant jusqu'à 5 ans Le temps de progression entre le premier évènement clinique et la SEPCD a été plus long dans le groupe où le traitement par COPAXONE a été instauré tôt par rapport au groupe ayant reçu le traitement plus tardivement, reflétant une réduction du risque de 41 % avec le traitement précoce versus le traitement retardé (risque estimé = 0,59, IC à 95 % [0,44-0,80], p = 0,0005). La proportion de sujets ayant progressé dans le groupe de traitement retardé était plus élevée (49,6 %) que dans le groupe de traitement précoce (32,9 %).
Un effet constant, en faveur d’un traitement précoce plutôt que d’un traitement retardé, a été montré sur le nombre annualisé de lésions sur l'ensemble de la période d'étude à la fois pour les nouvelles lésions T1 rehaussées par le Gd (réduction de 54 % ; p < 0,0001), les nouvelles lésions T2 (réduction de 42 % ; p < 0,0001), et les nouvelles lésions T1 hypo-intenses (réduction de 52 % ; p < 0,0001). Un effet en faveur du traitement précoce a également été observé sur les réductions du nombre total de nouvelles lésions T1 rehaussées par le Gd (réduction de 46 % ; p = 0,001), le volume de lésions T1 rehaussées par le Gd (différence moyenne de -0,06 mL ; p < 0,001), ainsi que le nombre total de nouvelles lésions T1 hypo-intenses (réduction de 46 %; p < 0,001) mesurées sur la totalité de la période de l'étude.
Aucune différence appréciable entre les cohortes de traitement précoce et de traitement retardé n'a été observée au niveau du volume des lésions T1 hypo-intenses ou de l'atrophie cérébrale pendant 5 ans. Cependant, l'analyse de l'atrophie cérébrale au moyen de la dernière valeur observée (ajustée en fonction de l'exposition au traitement) a montré une réduction en faveur du traitement précoce par AG (la différence moyenne de pourcentage de changement du volume cérébral était de 0,28 % ; p = 0,0209).
5.2. Propriétés pharmacocinétiques
Aucune étude de pharmacocinétique n'a été réalisée chez des patients. Les données in vitro et les données limitées provenant de volontaires sains indiquent qu'après l'administration sous-cutanée d'acétate de glatiramère, la substance active est facilement absorbée, et qu'une grande partie de la dose est rapidement dégradée en fragments plus petits, dès le tissu sous-cutané.
5.3. Données de sécurité préclinique
Un dépôt de complexe immun dans les glomérules rénaux a été rapporté chez un petit nombre de rats et de singes traités pendant au moins 6 mois. Dans une étude d'une durée de 2 ans chez le rat, il n'a pas été observé de dépôt de complexes immuns dans les glomérules rénaux.
Une réaction anaphylactique a été rapportée après administration à des animaux sensibilisés (cobaye et souris). La pertinence de ces résultats pour l'Homme est inconnue.
Une toxicité au site d'injection après administration répétée a été observée fréquemment chez l'animal.
Chez le rat, on a observé une réduction légère mais statistiquement significative du gain pondéral de la progéniture des femelles traitées pendant la grossesse et tout au long de l’allaitement aux doses sous-cutanées ≥ 6mg/kg/jour (2,83 fois la dose quotidienne maximale recommandée chez l’Homme pour un adulte de 60 kg sur la base de mg/m2) par rapport au groupe témoin. Aucun autre effet significatif sur la croissance de la progéniture et le développement du comportement n'a été observé.
Mannitol, eau pour préparations injectables.
3 ans.
6.4. Précautions particulières de conservation
Conserver les seringues préremplies dans l’emballage extérieur, à l’abri de la lumière.
A conserver au réfrigérateur (entre 2°C et 8°C).
Ne pas congeler.
Si les seringues préremplies ne peuvent pas être conservées au réfrigérateur, elles peuvent être conservées jusqu'à un mois maximum, entre 15°C et 25°C.
Une fois cette période d’un mois écoulée, si les seringues préremplies de COPAXONE 20 mg/ml n’ont pas été utilisées et si elles sont toujours dans leur emballage d’origine, elles doivent être remises au réfrigérateur (entre 2°C et 8°C).
6.5. Nature et contenu de l'emballage extérieur
Une seringue préremplie contenant une solution de COPAXONE pour injection se compose d'un corps de seringue de 1 ml en verre de type I transparent doté d'une aiguille fixe, d'un piston en polypropylène (polystyrène en option), d'un bouchon de piston en caoutchouc et d'un embout de protection pour l'aiguille.
Chaque seringue préremplie est emballée individuellement sous plaquette en PVC.
COPAXONE est disponible en boîtes de 7, 28 ou 30 seringues préremplies de 1 ml de solution injectable ou en conditionnement multiple de 90 (3 boîtes de 30) seringues préremplies de 1 ml de solution injectable.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Usage unique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
100-110 ESPLANADE DU GENERAL DE GAULLE
92931 PARIS LA DEFENSE CEDEX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 363 840 1 9 : 28 seringue(s) préremplie(s) en verre de 1 ml.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Prescription initiale et renouvellement réservés aux spécialistes en neurologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 17/11/2025
COPAXONE 20 mg/ml, solution injectable en seringue préremplie
Acétate de glatiramère
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que COPAXONE 20 mg/ml, solution injectable en seringue préremplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser COPAXONE 20 mg/ml, solution injectable en seringue préremplie ?
3. Comment utiliser COPAXONE 20 mg/ml, solution injectable en seringue préremplie ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver COPAXONE 20 mg/ml, solution injectable en seringue préremplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE COPAXONE 20 mg/ml, solution injectable en seringue préremplie ET DANS QUELS CAS EST-IL UTILISE ?
COPAXONE est un médicament indiqué dans le traitement des formes rémittentes de sclérose en plaques (SEP). Il modifie la manière dont fonctionne le système immunitaire de votre corps et il est classé comme agent immunomodulateur. On attribue les symptômes de la SEP à un dysfonctionnement du système immunitaire de l’organisme. Il en résulte une inflammation sous forme de plaques au niveau du cerveau et de la moelle épinière.
COPAXONE est utilisé pour réduire le nombre de poussées lors de la SEP (aggravations). Son efficacité n’a pas été démontrée si vous présentez une forme de SEP autre que rémittente et si vous ne présentez que peu ou pas de poussée. COPAXONE peut n’avoir aucun effet sur la durée ou sur la sévérité d’une poussée.
Il est indiqué pour le traitement de patients capables de marcher sans aide.
COPAXONE peut également être utilisé chez des patients ayant présenté pour la première fois, des symptômes évocateurs d'un risque élevé de développer une SEP. Avant de débuter le traitement, votre médecin devra éliminer toute autre cause médicale pouvant expliquer ces symptômes.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER COPAXONE 20 mg/ml, solution injectable en seringue préremplie ?
N’utilisez jamais COPAXONE 20 mg/ml, solution injectable en seringue préremplie :
· si vous êtes allergique à l'acétate de glatiramère ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
COPAXONE peut provoquer des réactions allergiques sévères qui peuvent parfois menacer le pronostic vital. Ces réactions peuvent survenir peu de temps après l’administration, mais également plusieurs mois voire plusieurs années après le début du traitement, et ce même si les précédentes administrations n’ont pas entraîné de réaction allergique.
Les signes et symptômes de réactions allergiques et de réactions post-injection peuvent être identiques. Votre médecin vous expliquera quels sont les signes d’une réaction allergique.
Adressez-vous à votre médecin ou pharmacien avant d'utiliser COPAXONE, si vous avez une insuffisance rénale ou des troubles cardiaques, car vous serez probablement soumis à des examens réguliers.
Adressez-vous à votre médecin ou pharmacien avant d'utiliser COPAXONE, si vous avez ou avez eu des troubles hépatiques (y compris ceux dus à la consommation d’alcool).
Enfants
COPAXONE ne doit pas être utilisé chez l'enfant de moins de 12 ans.
Personnes âgées
COPAXONE n'a pas été étudié chez le sujet âgé. Demandez conseil à votre médecin.
Autres médicaments et COPAXONE 20 mg/ml, solution injectable en seringue préremplie
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Si vous êtes enceinte, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil et avis auprès de votre médecin à propos d’un traitement par COPAXONE pendant la grossesse.
COPAXONE peut être utilisé pendant la grossesse sur avis de votre médecin.
Des données limitées chez l’être humain n’ont révélé aucun effet négatif de COPAXONE sur les nouveau-nés/nourrissons allaités. COPAXONE peut être utilisé pendant l’allaitement.
Conduite de véhicules et utilisation de machines
COPAXONE n'est pas connu pour influencer l'aptitude à conduire des véhicules ou à utiliser des machines.
3. COMMENT UTILISER COPAXONE 20 mg/ml, solution injectable en seringue préremplie ?
Chez les adultes et les adolescents de 12 ans et plus, la dose quotidienne recommandée est d’une seringue préremplie (20 mg d'acétate de glatiramère) administrée sous la peau (voie sous-cutanée).
Il est très important d’injecter correctement COPAXONE :
· Uniquement dans le tissu situé sous la peau (voie sous-cutanée) (voir « Mode d’emploi »).
· En respectant la dose indiquée par votre médecin. Utilisez uniquement la dose prescrite par votre médecin.
· N’utilisez jamais la même seringue plus d'une fois. Jetez tout produit inutilisé ou déchet.
· Ne mélangez pas ou n’administrez pas en même temps le contenu des seringues préremplies de COPAXONE avec un autre produit.
· Si la solution contient des particules, ne l'utilisez pas. Utilisez une nouvelle seringue.
La première fois que vous utiliserez COPAXONE vous recevrez une formation à la technique de l’auto-injection. Cette formation sera supervisée par un médecin ou un(e) infirmier/ère qui restera près de vous pendant que vous réaliserez votre première injection et pendant les 30 minutes suivant l'injection afin de s'assurer que vous n'avez aucun problème.
Mode d’emploi
Lisez attentivement ces instructions avant d'utiliser COPAXONE.
Avant de préparer l'injection, assurez-vous d'avoir tout le matériel nécessaire :
· Une seringue préremplie de COPAXONE dans son conditionnement individuel (plaquette).
· Le récipient destiné à la récupération des seringues et des aiguilles usagées.
· Pour chaque injection, ne sortez de la boîte qu'une seule plaquette contenant une seringue préremplie. Laissez les autres seringues dans la boîte.
· Si votre seringue a été conservée au réfrigérateur, sortez la plaquette contenant la seringue au moins 20 minutes avant votre injection, afin que la solution atteigne la température ambiante.
Lavez-vous soigneusement les mains à l'eau et au savon.
Si vous souhaitez utiliser un dispositif d’injection pour effectuer votre injection, le dispositif CSYNC peut être utilisé avec COPAXONE. Le dispositif CSYNC n’est approuvé que dans le cadre d’une utilisation avec COPAXONE et n’a pas été testé avec d’autres produits. Veuillez consulter le mode d'emploi fourni avec le CSYNC.
Choisissez un site d'injection se situant dans les zones, selon les schémas.
Vous disposez de sept zones possibles où injecter le médicament
· Zone 1 : Région de l’abdomen (ventre) autour du nombril. Evitez d’injecter à moins de 5 cm autour du nombril,
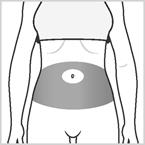
· Zone 2 et 3 : Cuisses (environ 5 cm au-dessus du genou et 5 cm au-dessous du pli de l’aine),

· Zone 4, 5, 6 et 7 : Arrière de la partie supérieure des bras (partie charnue supérieure arrière des bras) et de la partie supérieure des hanches (partie charnue supérieure de la hanche, toujours en-dessous de la taille),
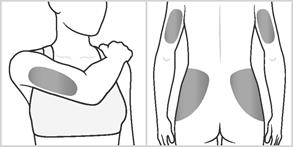
Chaque zone d'injection comporte plusieurs sites d'injection. Choisissez un site d'injection différent à chaque injection. Cela réduira le risque de réaction au site d’injection (irritation ou douleur). Alternez les zones d’injection et alternez également les sites d’injection dans une même zone. N'utilisez pas le même site à chaque fois.
Remarque : n’injectez pas à un endroit qui est douloureux ou décoloré ou qui comporte une induration. Il est recommandé de respecter un ordre planifié de rotation des sites d’injection et de noter les sites utilisés dans un agenda. Vous aurez peut-être des difficultés à injecter dans certaines zones de votre corps (comme la partie arrière de vos bras), et pourriez avoir besoin d’aide.
Comment réaliser l’injection :
· Retirez la seringue de son emballage de protection en décollant le film qui recouvre la plaquette.
· Retirez l'embout de protection de l'aiguille, ne retirez pas l’aiguille avec votre bouche ou avec vos dents.
· Pincez doucement la peau entre le pouce et l'index (figure 1).
· Insérez l'aiguille dans la peau de la manière décrite à la figure 2.
· Injectez le médicament en enfonçant complètement le piston de manière régulière, jusqu'à ce que la seringue soit vide.
· Enlevez d’une manière droite la seringue et l’aiguille.
· Jetez la seringue dans le récipient de récupération des déchets. Ne jetez pas les seringues usagées dans la poubelle de la maison mais dans le récipient spécifique résistant à la perforation, selon la recommandation de votre médecin ou votre infirmier/ère.
Figure 1 Figure 2

Si vous avez l'impression que l'effet de COPAXONE est trop fort ou trop faible, parlez-en à votre médecin.
Si vous avez utilisé plus de COPAXONE 20 mg/ml, solution injectable en seringue préremplie que vous n’auriez dû
Consultez immédiatement votre médecin.
Si vous oubliez d’utiliser COPAXONE 20 mg/ml, solution injectable en seringue préremplie
Si vous arrêtez d’utiliser COPAXONE 20 mg/ml, solution injectable en seringue préremplie
N’interrompez pas votre traitement par COPAXONE sans consulter votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Réactions allergiques (hypersensibilité, réaction anaphylactique)
Une réaction allergique grave peut se produire peu de temps après l’administration. Il s’agit d’un effet indésirable peu fréquent. Ces réactions peuvent également survenir plusieurs mois voire plusieurs années après le début du traitement par COPAXONE, même si les précédentes administrations n’ont pas entraîné de réaction allergique.
Arrêtez d'utiliser COPAXONE et prévenez immédiatement votre médecin ou rendez-vous dans le service médical d'urgence le plus proche, si vous constatez la survenue soudaine de l'un des effets indésirables suivants :
· éruption cutanée étendue (boutons rouges ou urticaire) ;
· gonflement des paupières, du visage, des lèvres, de la bouche, de la gorge ou de la langue ;
· difficultés pour respirer, essoufflement soudain ou respiration sifflante ;
· convulsions (crises) ;
· difficultés à avaler ou à parler ;
· syncope, étourdissement ou évanouissement ;
· état de choc.
Autres réactions suivant l'injection (réactions immédiates post-injection)
De façon peu fréquente, certaines personnes peuvent présenter un ou plusieurs des symptômes suivants, dans les minutes suivant l'injection de COPAXONE. Ces réactions sont généralement sans gravité et disparaissent habituellement au bout de 30 minutes.
Toutefois, si les symptômes suivants durent plus de 30 minutes, avertissez immédiatement votre médecin ou rendez-vous dans le service d'urgence de l’hôpital le plus proche :
· rougeur du torse ou du visage (vasodilatation) ;
· difficulté respiratoire (dyspnée) ;
· douleur dans la poitrine ;
· battements cardiaques rapides et forts (palpitations, tachycardie).
Troubles hépatiques
Des troubles hépatiques ou une aggravation des troubles hépatiques, y compris une insuffisance hépatique (certains cas nécessitant une transplantation hépatique), peuvent rarement se produire avec COPAXONE. Contactez immédiatement votre médecin si vous présentez des symptômes, tels que :
· nausées ;
· perte d'appétit ;
· urine de couleur foncée et selles pâles ;
· jaunissement de la peau ou de la partie blanche de l’œil ;
· saignements apparaissant plus facilement que d'habitude.
Les effets indésirables suivants ont été signalés avec COPAXONE :
Très fréquent : pouvant affecter plus de 1 personne sur 10
· infection, grippe ;
· anxiété, dépression ;
· maux de tête ;
· nausées ;
· éruption cutanée ;
· douleur des articulations ou du dos ;
· sensation de faiblesse, réactions cutanées au site d'injection, telles que rougeur, douleur, formation de papules, démangeaisons, gonflement des tissus, inflammation et hypersensibilité au site d'injection (ces réactions au site d'injection ne sont pas inhabituelles et diminuent généralement avec le temps), douleur non spécifique.
Fréquent : pouvant affecter jusqu'à 1 personne sur 10
· inflammation des voies respiratoires, grippe intestinale, bouton de fièvre, otite, nez qui coule, abcès dentaires, mycose vaginale ;
· tumeur cutanée non maligne (néoplasme non malin de la peau), tumeur des tissus (néoplasme) ;
· gonflement des ganglions ;
· réactions allergiques ;
· prise de poids, perte d'appétit ;
· nervosité ;
· altération du goût, hypertonicité musculaire, migraine, problèmes d'élocution, syncope, tremblements ;
· problèmes oculaires, vision double ;
· affection de l’oreille ;
· toux, rhinite allergique ;
· problèmes au niveau de l’anus ou du rectum, constipation, caries dentaires, indigestion, difficultés à avaler, incontinence fécale, vomissements ;
· anomalie des tests de la fonction hépatique ;
· ecchymoses, transpiration excessive, démangeaisons, urticaire et autres problèmes cutanés ;
· douleur dans le cou ;
· besoin d’uriner impérieux ou fréquent, incapacité à vider complètement votre vessie ;
· frissons, gonflement du visage, amincissement du tissu sous-cutané au site d’injection, réaction locale, œdème périphérique, fièvre.
Peu fréquent : pouvant affecter jusqu'à 1 personne sur 100
· abcès, inflammation de la peau et des tissus mous sous-cutanés, furoncles, zona, inflammation des reins ;
· cancer de la peau ;
· modification du nombre de globules blancs (augmentation ou réduction), augmentation du volume de la rate, diminution du nombre de plaquettes, modification de la forme des globules blancs ;
· augmentation de volume ou hyperactivité de la thyroïde ;
· faible tolérance à l'alcool, goutte, augmentation des graisses (lipides) dans le sang, augmentation du taux de sodium dans le sang, diminution de la ferritine dans le sang ;
· rêves étranges, confusion, euphorie, hallucinations (voir, entendre, sentir, gouter ou ressentir des choses qui n’existent pas), agressivité, modification de la personnalité, altération de l’humeur, tentative de suicide ;
· engourdissement et douleur de la main (syndrome du canal carpien), troubles mentaux, crises (convulsions), troubles de l'écriture et de la lecture, troubles musculaires, troubles du mouvement, spasmes musculaires, inflammation des nerfs, anomalie des connections neuromusculaires entraînant un fonctionnement musculaire anormal, mouvements involontaires rapides des yeux, paralysie, impossibilité de relever le pied (sciatique paralysante), état inconscient (stupeur), déficit du champ visuel ;
· cataracte, lésion de la cornée, sécheresse oculaire, saignement oculaire, paupière tombante, dilatation de la pupille, atteinte du nerf optique entraînant des troubles de la vue ;
· augmentation des battements cardiaques, battements cardiaques lents ou rapides ;
· varices ;
· apnée, saignement de nez, respiration anormalement rapide ou profonde (hyperventilation), sensation de gorge serrée, troubles pulmonaires, sensation d’étouffement ;
· inflammation du côlon ou de l’intestin, polypes du côlon, éructations, ulcère de l'œsophage, inflammation des gencives, saignement rectal, augmentation de volume des glandes salivaires ;
· calculs biliaires, augmentation du volume du foie ;
· gonflement de la peau et des tissus mous, éruption cutanée de contact, nodules cutanés, nodules cutanés rouges et douloureux ;
· gonflement, inflammation et douleur des articulations (arthrite ou ostéoarthrite), inflammation du liquide synovial (existant dans certaines articulations) et douleurs, douleur au flanc, diminution de la masse musculaire ;
· sang dans les urines, calculs rénaux, affection des voies urinaires, urines anormales ;
· gonflement des seins, difficultés à obtenir une érection, descente d’organes (prolapsus), érection prolongée, troubles vaginaux, de la prostate ou des testicules, saignement vaginal, frottis vaginal anormal ;
· kyste, sensation de gueule de bois, température corporelle inférieure à la normale (hypothermie), inflammation, destruction des tissus au site d'injection, problèmes au niveau des muqueuses ;
· troubles après une vaccination.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER COPAXONE 20 mg/ml, solution injectable en seringue préremplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et la boîte (EXP). La date de péremption fait référence au dernier jour de ce mois.
A conserver au réfrigérateur (entre 2°C et 8°C).
Les seringues préremplies de COPAXONE peuvent être conservées en dehors du réfrigérateur, jusqu'à un mois maximum, entre 15°C et 25°C. Cela ne peut être fait qu’une seule fois. Après un mois, toute seringue préremplie de COPAXONE n’ayant pas été utilisée et toujours dans son emballage d’origine doit être remise au réfrigérateur.
Ne pas congeler.
Conserver les seringues préremplies dans l’emballage extérieur, à l’abri de la lumière.
Jeter toute seringue préremplie présentant des particules.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient COPAXONE 20 mg/ml, solution injectable en seringue préremplie
· La substance active est :
Glatiramère............................................................................................................................ 18 mg
Sous forme d’acétate de glatiramère....................................................................................... 20 mg
Pour 1 seringue préremplie (1 ml).
· Les autres composants sont :
Mannitol, eau pour préparations injectables.
COPAXONE, solution injectable en seringue préremplie se présente sous forme d’une solution injectable, limpide, exempte de particules visibles.
Chaque seringue préremplie est emballée individuellement sous plaquette en PVC.
COPAXONE est disponible en boîtes de 7, 28 ou 30 seringues préremplies de 1 ml de solution injectable ou en conditionnement multiple de 90 seringues préremplies comprenant 3 boîtes contenant chacune 30 seringues préremplies de 1 ml de solution injectable.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
100-110 ESPLANADE DU GENERAL DE GAULLE
92931 PARIS LA DEFENSE CEDEX
Exploitant de l’autorisation de mise sur le marché
100-110 ESPLANADE DU GENERAL DE GAULLE
NORTON HEALTHCARE LIMITED T/A IVAX PHARMACEUTICALS UK (TEVA RUNCORN)
ASTON LANE NORTH, WHITEHOUSE VALE INDUSTRIAL ESTATE
RUNCORN, CHESHIRE, WA7 3FA
ROYAUME-UNI
OU
ACTAVIS GROUP PTC EHF.
DALSHRAUN 1
220, HAFNARFJOROUR
ISLANDE
OU
MERCKLE GMBH
GRAF-ARCO-STR.3
89079 ULM
Allemagne
Noms du médicament dans les Etats membres de l'Espace Economique Européen
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).