Dernière mise à jour le 03/08/2026
CABAZITAXEL REDDY PHARMA 60 mg, solution à diluer et solvant pour solution pour perfusion
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique : Agent antinéoplasiques. Code ATC : L01CD04.
CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion est utilisé pour traiter le cancer de la prostate qui a progressé après avoir eu une autre chimiothérapie. Il fonctionne en stoppant la croissance et la multiplication des cellules.
Vous prendrez aussi un autre médicament qui fait partie de votre traitement. Il s’agit d’un corticostéroïde (prednisone ou prednisolone) par voie orale tous les jours. Demandez à votre médecin qu’il vous donne des informations concernant cet autre médicament.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 22/07/2020 | Inscription (CT) | Le service médical rendu par CABAZITAXEL REDDY PHARMA 60 mg, solution à diluer et solvant pour solution pour perfusion est important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 22/07/2020 | Inscription (CT) | Cette spécialité est un générique qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à la spécialité de référence, JEVTANA 60 mg, solution à diluer et solvant pour solution pour perfusion, déjà inscrite. |
ANSM - Mis à jour le : 06/09/2022
CABAZITAXEL REDDY PHARMA 60 mg, solution à diluer et solvant pour solution pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Cabazitaxel............................................................................................................................ 40 mg
Pour 1 ml de solution à diluer.
Chaque flacon de 1,5 ml (volume nominal) de solution à diluer contient 60 mg de cabazitaxel.
Après dilution initiale avec la totalité du solvant, chaque ml de la solution contient 10 mg de cabazitaxel.
Remarque : le flacon de solution à diluer de CABAZITAXEL REDDY PHARMA 60 mg/1,5 ml (volume de remplissage : 73,2 mg de cabazitaxel/1,83 ml) et le flacon de solvant (volume de remplissage : 5,70 ml) contiennent tous les deux un surremplissage pour compenser la perte de liquide survenant lors de la préparation. Ce surremplissage permet d’obtenir, après dilution avec la TOTALITE du contenu du flacon de solvant fourni, une solution contenant 10 mg/ml de cabazitaxel.
Excipient à effet notoire : chaque flacon de solvant contient 573,3 mg d’éthanol à 96%.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution à diluer et solvant pour solution pour perfusion (solution à diluer stérile).
La solution à diluer est une solution huileuse, limpide, jaune à jaune marron.
Le solvant est une solution limpide et légèrement jaune.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Prémédication
La prémédication recommandée doit être faite au moins 30 minutes avant chaque administration de CABAZITAXEL REDDY PHARMA avec les médicaments injectés par voie intraveineuse suivants afin de diminuer le risque et la sévérité de l’hypersensibilité :
· Antihistaminique (dexchlorphéniramine 5 mg ou diphénydramine 25 mg ou équivalent),
· Corticostéroïde (dexaméthasone 8 mg ou équivalent), et
· Antagonistes H2 (ranitidine ou équivalent) (cf. rubrique 4.4).
Une prophylaxie antiémétique est recommandée et peut être donnée oralement ou par voie intraveineuse si besoin.
Au cours du traitement, une hydratation efficace du patient doit être garantie, pour prévenir des complications comme une insuffisance rénale.
Posologie
La posologie recommandée de mg/m REDDY PHARMA est 25 mg/m2 administrée par perfusion de 1 heure toutes les 3 semaines en association avec 10 mg par jour de prednisone ou prednisolone administrée par voie orale pendant tout le traitement.
Ajustement des doses
Une modification de la dose doit être faite chez les patients ayant présenté les effets indésirables suivants (les Grades font référence aux Critères de la Terminologie Commune des Effets Indésirables [CTCAE 4.0]) :
Tableau 1 – Modification de dose recommandée pour effets indésirables chez des patients traités avec cabazitaxel
|
Effet indésirable |
Modification de la dose |
|
Neutropénie prolongée de grade ≥3 (de plus d’une semaine) malgré un traitement approprié incluant du G-CSF
|
Reporter le traitement jusqu’à ce que le nombre de neutrophiles soit >1 500 cellules/mm3, puis réduire la dose de cabazitaxel de 25 mg/m2 à 20 mg/m2. |
|
Neutropénie fébrile ou infection neutropénique |
Reporter le traitement jusqu’à amélioration ou normalisation, et jusqu’à ce que le nombre de neutrophiles soit >1 500 cellules/mm3, puis réduire la dose de cabazitaxel de 25 mg/m2 à 20 mg/m2. |
|
Diarrhée de grade ≥3 ou diarrhées persistantes malgré un traitement approprié, incluant solutés et électrolytes de substitution
|
Retarder le traitement jusqu’à amélioration ou normalisation, puis réduire la dose de cabazitaxel de 25 mg/m2 à 20 mg/m2. |
|
Neutropénie périphérique de grade >2 |
Retarder le traitement jusqu’à amélioration puis réduire la dose de cabazitaxel de 25 mg/m2 à 20 mg/m2. |
Si les patients continuent à présenter l’un de ces effets indésirables à 20 mg/m2, une réduction supplémentaire à 15 mg/m2 ou un arrêt de CABAZITAXEL REDDY PHARMA doit être envisagé. Les données chez les patients à une dose inférieures à 20 mg/m2 sont limitées.
Populations particulières
Patients avec insuffisance hépatique
Le cabazitaxel est largement métabolisé par le foie. Les patients ayant une insuffisance hépatique légère (bilirubine totale >1 à ≤ 1,5 x la limite supérieure de la normale (LSN) ou Aspartate Aminotransférase (ASAT) > 1,5 x LSN), doivent recevoir une dose réduite de cabazitaxel de 20 mg/m2. L’administration de cabazitaxel chez des patients ayant une insuffisance hépatique légère doit être effectuée avec une attention particulière et la tolérance doit être surveillée étroitement.
Le cabazitaxel ne doit pas être administré à des patients d’insuffisance hépatique modérée et sévère (bilirubine totale >1.5 x LSN) (voir rubriques 4.3, 4.4 et 5.2).
Patient avec insuffisance rénale
Le cabazitaxel est très peu excrété par le rein. Aucun ajustement posologique n’est nécessaire chez des patients ayant une insuffisance rénale, ne nécessitant pas une hémodialyse. Compte-tenu de l’état des patients présentant une maladie rénale en phase terminale (clairance de la créatinine (CLCR< 15mL/min/1,73 m2) et des données disponibles limitées, ces patients doivent être traités avec précaution et suivis étroitement pendant le traitement (voir rubriques 4.4 et 5.2).
Patients âgés
Aucun ajustement posologique spécifique n’est recommandé pour l’utilisation du cabazitaxel chez les patients âgés (voir également rubriques 4.4, 4.8 et 5.2).
Prise concomitante de médicaments
La prise concomitante de médicaments qui sont de puissants inducteurs ou de puissants inhibiteurs du CYP3A doit être évitée.
Cependant, si certains patients nécessitent la co-administration d’un puissant inhibiteur du CYP3A, une réduction de la dose de cabazitaxel de 25% devra être envisagée (voir rubrique 4.4 et 4.5).
Population pédiatrique
Il n’y a pas d’utilisation justifiée de CABAZITAXEL REDDY PHARMA dans la population pédiatrique.
La sécurité et l’efficacité de CABAZITAXEL REDDY PHARMA chez les enfants et les adolescents de moins de 18 ans n’ont pas été établies (voir rubrique 5.1).
Mode d’administration
Pour les instructions de préparation et d’administration du médicament, voir la rubrique 6.6.
Les poches de perfusion en PVC et les sets de perfusion en polyuréthane ne doivent pas être utilisées. CABAZITAXEL REDDY PHARMA ne doit pas être mélangé avec des médicaments autres que ceux mentionnés à la rubrique 6.6.
· Nombre de neutrophiles inférieur à 1 500/mm3.
· Insuffisance hépatique modérée et sévère (bilirubine totale> 3 x LSN).
· Vaccination concomitante avec le vaccin contre la fièvre jaune (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
Tous les patients doivent recevoir une prémédication avant l’initiation de la perfusion de cabazitaxel (voir rubrique 4.2).
Les patients doivent être étroitement surveillés pour les réactions d’hypersensibilité, essentiellement pendant la première et la seconde perfusion. Les réactions d’hypersensibilité peuvent survenir dans les quelques minutes suivant l’initiation de la perfusion de cabazitaxel, ainsi les installations et équipements pour le traitement de l’hypotension et bronchospasmes devraient être à proximité du patient. Des réactions sévères peuvent survenir, incluant rash/érythèmes généralisés, hypotension et bronchospasmes. Les réactions d’hypersensibilité sévères nécessitent un arrêt immédiat du cabazitaxel et un traitement approprié. Les patients avec une réaction d’hypersensibilité doivent arrêter le traitement par CABAZITAXEL REDDY PHARMA (voir rubrique 4.3).
Myélosuppression
Une myélosuppression se manifestant par une neutropénie, anémie, thrombopénie, ou pancytopénie (voir « Risques de neutropénies » et « Anémie » en rubrique 4.4 ci-dessous) peut survenir.
Risque de neutropénies
Les patients traités par cabazitaxel peuvent recevoir une prophylaxie par G-CSF, conformément aux guidelines de l’American Society of Clinical Oncology (ASCO) et/ou recommandations institutionnelles en vigueur, pour réduire le risque ou prendre en charge les complications des neutropénies (neutropénies fébriles, neutropénies prolongées ou infections neutropéniques). Une prophylaxie primaire avec G-CSF doit être considérée chez les patients ayant des facteurs de risque clinique important (âge > 65 ans, mauvais état général, épisodes précédents de neutropénie fébrile, champ d’irradiation antérieur extensif, mauvais état nutritionnel, ou d’autres facteurs de comorbidités sévères) qui les prédisposent à une augmentation des complications liées à une neutropénie prolongée.
L’utilisation de G-CSF a montré qu’elle limitait l’incidence et la sévérité des neutropénies.
La neutropénie est l’effet indésirable le plus fréquent du cabazitaxel (voir rubrique 4.8). Le suivi de la numération formule sanguine hebdomadaire est essentiel pendant le cycle 1 et ensuite avant chaque cycle de traitement, afin d’ajuster la dose si besoin.
La dose doit être réduite en cas de neutropénie fébrile, ou neutropénie prolongée malgré un traitement approprié (voir rubrique 4.2).
Le traitement ne devra être repris chez ces patients seulement quand les neutrophiles seront ≥ 1 500/mm3 (voir rubrique 4.3).
Affections gastro-intestinales
Des symptômes tels que des douleurs et une sensibilité abdominales, fièvre, constipation persistante, diarrhée, avec ou sans neutropénie, peuvent être des manifestations précoces d’une toxicité gastro-intestinale et doivent être évalués et traités rapidement. Le traitement par cabazitaxel peut alors être repoussé ou arrêté si nécessaire.
Risque de nausée, vomissement, diarrhée et déshydratation
Si des patients ont eu des antécédents de diarrhées après une administration de cabazitaxel, ils doivent être traités par des médicaments anti-diarrhéiques habituellement utilisés. Des mesures appropriées doivent être prises pour réhydrater ces patients. Des diarrhées peuvent se produire plus fréquemment chez des patients ayant reçu une irradiation abdomino-pelvienne. Une déshydratation est plus fréquente chez les patients âgés de 65 ans ou plus. Des mesures appropriées doivent être prises pour réhydrater les patients, les suivre et corriger leur taux sérique d’électrolytes notamment le potassium. Un report du traitement ou une réduction de la dose peuvent être nécessaires pour des diarrhées de grade ≥ 3 (voir rubrique 4.2). Si des patients ont eu des antécédents de nausées et vomissements, ils doivent être traités avec des antiémétiques habituellement utilisés.
Risque de réactions gastro-intestinales graves
Des hémorragies et des perforations digestives, des iléus, des colites, incluant des issues fatales ont été rapportées chez des patients traités par cabazitaxel (voir rubrique 4.8). Une attention est requise chez les patients les plus à risque de développer des complications digestives : ceux souffrant de neutropénie, les patients âgés, en cas d’utilisation concomitante d’AINS, d’antiagrégants plaquettaires ou d’anticoagulants, et chez les patients ayant été antérieurement traités par radiothérapie pelvienne ou présentant des antécédents digestifs, comme des ulcérations ou des saignements digestifs.
Neuropathie périphérique
Des cas de neuropathie périphérique, de neuropathie sensitive périphérique (par exemple, les paresthésies, dysesthésies) et de neuropathie périphérique motrice ont été observés chez les patients recevant du cabazitaxel. Les patients sous traitement par cabazitaxel doivent informer leur médecin avant de poursuivre le traitement si les symptômes de neuropathie apparaissent, tels qu’une douleur, une brûlure, un picotement, un engourdissement ou une faiblesse. Les médecins doivent évaluer la présence ou l’aggravation de la neuropathie avant chaque traitement. Le traitement doit être retardé jusqu’à amélioration des symptômes. La dose de cabazitaxel doit être réduite de 25 mg/m2 à 20 mg/m2 devant une neuropathie périphérique de grade ≥ 2 persistante (voir rubrique 4.2).
Anémie
Des anémies ont été observées chez les patients recevant du cabazitaxel (voir rubrique 4.8). Le taux d’hémoglobine et l’hématocrite doivent être contrôlés avant le traitement par cabazitaxel ainsi que lorsque les patients présentent des signes ou symptômes d’anémie ou de perte de sang. Une attention particulière est recommandée chez les patients ayant une hémoglobine < 10 g/dL et des mesures appropriées devront être prises en fonction de la cliniques.
Risque d’insuffisance rénale
Des troubles rénaux associés à des infections, des déshydratations sévères dues à des diarrhées, des vomissements et des uropathies obstructives ont été rapportés.
Des insuffisances rénales incluant des cas avec une issue fatale ont été observées. Des mesures appropriées doivent être prises pour en identifier la cause et traiter les patients.
Une hydratation adéquate doit être assurée tout au long du traitement par cabazitaxel. Le patient être informé de la nécessité de signaler immédiatement tout changement de diurèse quotidienne. La créatinine plasmatique devra être mesurée à l’initiation, à chaque bilan sanguin, et chaque fois que le patient rapporte une modification de sa diurèse. Le traitement par cabazitaxel doit être interrompu en cas de dégradation de la fonction rénale conduisant à une insuffisance rénale de Grade ≥ 3 du CTCAE 4.0.
Affections respiratoires
Des pneumonies interstitielles/pneumopathies inflammatoires et des pneumopathies interstitielles diffuses ont été rapportées et peuvent être associées à une issue fatale (voir rubrique 4.8).
Si de nouveaux symptômes pulmonaires apparaissent ou des symptômes pulmonaires s’aggravent, les patients doivent être surveillés de façon rapprochée, rapidement examinés, et traités de façon appropriée.
L’interruption de traitement par cabazitaxel est recommandée jusqu’à ce que le diagnostic soit établi. Une prise en charge précoce peut aider à améliorer l’état du patient. Le bénéfice de la reprise du traitement par cabazitaxel doit être évalué avec attention.
Risque d’arythmie cardiaque
Des arythmies cardiaques ont été rapportées, plus fréquemment des tachycardies et des fibrillations auriculaires (voir rubrique 4.8).
Patients âgés
Les patients âgés (≥ 65 ans) peuvent être plus susceptibles de présenter des effets indésirables incluant des neutropénies et des neutropénies fébriles (voir rubrique 4.8).
Patients avec insuffisance hépatique
Le traitement par cabazitaxel est contre-indiqué chez les patients ayant une insuffisance hépatique modérée et sévère (bilirubine totale > 1,5 x LSN) (voir rubriques 4.3 et 5.2).
La dose doit être réduite pour les patients ayant une insuffisance hépatique légère (bilirubine totale > 1 à 1,5 x LSN ou ASAT > 1,5 x LSN) (voir rubriques 4.2 et 5.2).
Interactions
La co-administration d’inhibiteurs puissants du CYP3A doit être évitée car ils peuvent augmenter les concentrations plasmatiques du cabazitaxel (voir rubriques 4.2 et 4.5). Si la co-administration avec un puissant inhibiteur du CYP3A ne peut pas être évitée, une surveillance étroite de la toxicité et une réduction de la dose du cabazitaxel devront être envisagées (voir rubriques 4.2 et 4.5). La co-administration d’inducteurs puissants du CYP3A doit être évitée car ils peuvent diminuer les concentrations plasmatiques du cabazitaxel (voir rubriques 4.2 et 4.5).
Excipients
Ce médicament contient 573 mg d’alcool (éthanol) dans chaque flacon de solvant. La quantité contenue dans la dose de ce médicament est équivalente à moins de 15 ml de bière ou 6 ml de vin. La faible quantité d’alcool dans ce médicament n’aura pas d’effet notable.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Inhibiteurs du CYP3A
L’administration répétée de kétoconazole (400 mg une fois par jour), un inhibiteur puissant du CYP3A, a conduit à une diminution de la clairance du cabazitaxel de 20% correspondant à une augmentation de l’AUC (aire sous la courbe) de 25%. En conséquence l’administration concomitante d’inhibiteurs puissants du CYP3A (tels que kétoconazole, itraconazole, clarithromycine, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycine, voriconazole) devra être évitée étant donné qu’une augmentation des concentrations plasmatiques du cabazitaxel peut survenir (voir rubriques 4.2 et 4.4).
L’administration concomitante d’aprepitant, un inhibiteur modéré du CYP3A, n’a pas eu d’effet sur la clairance du cabazitaxel.
Inducteurs du CYP3A
L’administration répétée de rifampicine (600 mg une fois par jour), un inducteur puissant du CYP3A, a conduit à une augmentation de la clairance du cabazitaxel de 21% correspondant à une diminution de l’AUC (aire sous la courbe) de 17%. En conséquence l’administration concomitante d’inducteurs puissants du CYP3A (tels que phénytoïne, carbamazépine, rifampicine, rifabutine, rifapentine, phénobarbital) devra être évitée étant donné qu’une diminution des concentrations plasmatiques du cabazitaxel peut survenir (voir rubriques 4.2 et 4.4). De plus, les patients doivent aussi s’abstenir de prendre du millepertuis.
OATP1B1
In vitro, il a également été montré que le cabazitaxel inhibe les protéines de transport OATP1B1 (polypeptides transporteurs d’anions organiques). Le risque d’interaction avec les substrats des OATP1B1 (par exemple les statines, le valsartan, le répaglinide) existe notamment pendant la durée de la perfusion (1 heure) et jusqu’à 20 minutes après la fin de la perfusion.
Il est recommandé de respecter un intervalle de 12 heures avant la perfusion et d’au moins 3 heures après la fin de la perfusion avant d’administrer des substrats des OATP1 B1.
Vaccinations
L’administration de vaccins vivants ou atténués chez des patients immunodéprimés par des agents de chimiothérapie peut entraîner des infections sévères ou fatales. La vaccination avec des vaccins vivants doit être évitée chez les patients recevant du cabazitaxel. Les vaccins tués ou inactifs peuvent être administrés : cependant la réponse de tels vaccins pourra être diminuée.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n’y a pas de données sur l’utilisation du cabazitaxel chez la femme enceinte. Des études chez des animaux ont montré une toxicité sur la reproduction à des doses maternotoxiques (voir rubrique 5.3) et un passage de la barrière placentaire par le cabazitaxel (voir rubrique 5.3). Comme tous les autres produits cytotoxiques, le cabazitaxel peut nuire au fœtus chez les femmes enceintes exposées. Le cabazitaxel n’est pas recommandé pendant la grossesse et chez les femmes n’utilisant pas de contraception.
Des données pharmacocinétiques disponibles chez l’animal ont montré une excrétion du cabazitaxel et de ses métabolites dans le lait maternel (voir rubrique 5.3). Un risque chez les enfants qui sont allaités ne peut être exclu.
Le cabazitaxel ne doit pas être utilisé pendant l’allaitement.
Fertilité
Des études chez l’animal ont montré que le cabazitaxel affectait le système de reproduction chez les rats mâles et les chiens sans aucun effet fonctionnel sur la fertilité (voir rubrique 5.3). Toutefois, considérant l’activité pharmacologique des taxanes, le potentiel génotoxique et l’effet de plusieurs composés de cette classe sur la fertilité dans les études animales, l’effet sur la fertilité masculine ne peut être exclu chez l’homme.
Compte tenu des effets potentiels sur les gamètes mâles et l’exposition potentielle dans le liquide séminal, les hommes traités par cabazitaxel doivent utiliser une contraception efficace pendant tout le traitement. Il est recommandé de poursuivre cette contraception jusqu’à 6 mois après la dernière dose de cabazitaxel.
En raison de l’exposition potentielle dans le liquide séminal, les hommes traités par cabazitaxel doivent éviter tout contact de leur sperme avec une autre personne au cours du traitement. Les hommes traités par cabazitaxel sont invités à demander des conseils sur la conservation du sperme avant traitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Il est recommandé aux patients ayant eu des antécédents de ces effets indésirables pendant le traitement de ne pas conduire ou d’utiliser des machines.
Résumé du profil de tolérance
La tolérance du cabazitaxel en association avec prednisone ou prednisolone a été évaluée dans 3 études randomisées, en ouvert, contrôlées (TROPIC, PROSELICA et CARD) chez un total de 1092 patients ayant un cancer de la prostate métastatique résistant à la castration qui ont été traités par cabazitaxel à 25 mg/m² une fois toutes les 3 semaines. Les patients ont reçu une médiane de 6 cycles de cabazitaxel.
Les incidences de l’analyse poolée des 3 études sont présentées ci-dessous et dans le tableau.
Les effets indésirables les plus fréquents (≥ 10%) quel que soit le grade étaient l’anémie (99,0%), les leucopénies (93,0%), les neutropénies (87,9%), les thrombopénies (41,1%), les diarrhées (42,1%), la fatigue (25,0%) et l’asthénie (15,4%).
Les effets indésirables les plus fréquents de grade ≥3 survenant chez au moins 5% des patients étaient les neutropénies (73,1%), les leucopénies (59,5%), l’anémie (12,0%), les neutropénies fébriles (8,0%), et les diarrhées (4,7%) .
L’arrêt du traitement lié aux effets indésirables est survenu à des fréquences similaires dans les 3 études (18,3% dans TROPIC, 19,5% dans PROSELICA et 19,8% dans CARD) chez les patients recevant du cabazitaxel. Les effets indésirables les plus fréquents (> 1.0%) entraînant l’arrêt du cabazitaxel étaient l’hématurie, la fatigue et les neutropénies.
Tableau des effets indésirables
Les effets indésirables sont listés dans le tableau 2 selon le système des classes de systèmes d’organe MedDRA et les catégories de fréquence. Au sein de chaque groupe de fréquence, les effets indésirables sont présentés selon un ordre décroissant de gravité. L’intensité des effets indésirables est gradée selon le CTCAE 4.0 (grade ≥ 3 = G ≥ 3). Les fréquences sont basées sur tous les grades et définies comme : très fréquent (≥ 1/10), fréquent (≥ 1/100 à <1/10), peu fréquent (≥ 1/1000 à <1/100), rare (≥ 1/10000 à <1/1000), très rare (<1/10000), inconnu (ne peut être estimé à partir des données disponibles).
Tableau 2 : Les effets indésirables rapportés et anomalies hématologiques avec cabazitaxel en association avec prednisone ou prednisolone de l’analyse poolée (n = 1092)
|
Classe de système d’organes |
Effets indésirables |
Tous grades n (%) |
Grade>3 n (%) |
||
|
|
|
Très fréquents |
Fréquents |
Peu fréquent |
|
|
Infections et infestations |
Infection neutropénique/ état septique |
|
48 (4,4) |
|
42 (3,8) |
|
Choc septique |
|
|
10 (0,9) |
10 (0,9) |
|
|
Sepsis |
|
13 (1,2) |
|
13 (1,2) |
|
|
Cellulite |
|
|
8 (0,7) |
3 (0,3) |
|
|
Infection du tractus urinaire |
|
103 (9,4) |
|
19 (1,7) |
|
|
Syndrome grippal |
|
22 (2,0) |
|
0 |
|
|
Cystite |
|
22 (2,0) |
|
2 (0,2) |
|
|
Infection des voies respiratoires supérieures |
|
23 (2,1) |
|
0 |
|
|
Herpès Zona |
|
14 (1,3) |
|
0 |
|
|
Candidose |
|
11 (1,0) |
|
1 (<0,1) |
|
|
Affections hématologiques et du système lymphatique |
Neutropéniea* |
950 (87,9) |
|
|
790 (73,1) |
|
Anémiea |
1073 (99,0) |
|
|
130 (12,0) |
|
|
Leukopeniaa |
1008 (93,0) |
|
|
645 (59,5) |
|
|
Thrombocytopéniea |
478 (44,1) |
|
|
44 (4,1) |
|
|
Neutropénie fébrile |
|
87 (8,0) |
|
87 (8,0) |
|
|
Affections du système immunitaire |
Hypersensitivité |
|
|
7 (0,6) |
0 |
|
Troubles du métabolisme et de la nutrition |
Anorexie |
192 (17,6) |
|
|
11 (1,0) |
|
Déshydratation |
|
27 (2,5) |
|
11 (1,0) |
|
|
Hyperglycémie |
|
11 (1,0) |
|
7 (0,6) |
|
|
Hypokalémie |
|
|
8 (0,7) |
2 (0,2) |
|
|
Affections psychiatriques |
Insomnie |
|
45 (4,1) |
|
0 |
|
Anxiété |
|
13 (1,2) |
|
0 |
|
|
Etat confusionnel |
|
12 (1,1) |
|
2 (0,2) |
|
|
Affections du système nerveux |
Dysgueusie |
|
64 (5,9) |
|
0 |
|
Trouble du goût |
|
56 (5,1) |
|
0 |
|
|
Neuropathie périphérique |
|
40 (3,7) |
|
2 (0,2) |
|
|
Neuropathie périphérique sensorielle |
|
89 (8,2) |
|
6 (0,5) |
|
|
Polyneuropathie |
|
|
9 (0,8) |
2 (0,2) |
|
|
Paresthésie |
|
46 (4,2) |
|
0 |
|
|
Hypoesthésie |
|
18 (1,6) |
|
1 (<0,1) |
|
|
Vertige |
|
63 (5,8) |
|
0 |
|
|
Céphalées |
|
56 (5,1) |
|
1 (<0,1) |
|
|
Léthargie |
|
15 (1,4) |
|
1 (<0,1) |
|
|
Sciatique |
|
|
9 (0,8) |
1 (<0,1) |
|
|
Affections oculaires |
Conjonctivites |
|
11 (1,0) |
|
0 |
|
Larmoiement augmenté |
|
22 (2,0) |
|
0 |
|
|
Affections des oreilles et du labyrinthe |
Acouphène |
|
|
7 (0,6) |
0 |
|
Vertige |
|
15 (1,4) |
|
1 (<0,1) |
|
|
Affections cardiaques* |
Fibrillation auriculaire |
|
14 (1,3) |
|
5 (0,5) |
|
Tachycardie |
|
11 (1,0) |
|
1 (<0,1) |
|
|
Affections vasculaires |
Hypotension |
|
38 (3,5) |
|
5 (0,5) |
|
Thrombose veineuse profonde |
|
12 (1,1) |
|
9 (0,8) |
|
|
Hypertension |
|
29 (2,7) |
|
12 (1,1) |
|
|
Hypotension orthostatique |
|
|
6 (0,5) |
1 (<0,1) |
|
|
Bouffée de chaleur |
|
23 (2,1) |
|
1 (<0,1) |
|
|
Flushing |
|
|
9 (0,8) |
0 |
|
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée |
|
97 (8,9) |
|
9 (0,8) |
|
Toux |
|
79 (7,2) |
|
0 |
|
|
Douleur oropharyngée |
|
26 (2,4) |
|
1 (< 0,1) |
|
|
Pneumonie |
|
26 (2,4) |
|
16 (1,5) |
|
|
Embolie pulmonaire |
|
30 (2,7) |
|
23 (2,1) |
|
|
Affections gastro-intestinales |
Diarrhée |
460 (42,1) |
|
|
51 (4,7) |
|
Nausée |
347 (31,8) |
|
|
14 (1,3) |
|
|
Vomissement |
207 (19,0) |
|
|
14 (1,3) |
|
|
Constipation |
202 (18,5) |
|
|
8 (0,7) |
|
|
Douleur abdominale |
|
105 (9,6) |
|
15 (1,4) |
|
|
Dyspepsie |
|
53 (4,9) |
|
0 |
|
|
Douleur abdominale haute |
|
46 (4,2) |
|
1 (< 0,1) |
|
|
Hémorrhoïdes |
|
22 (2,0) |
|
0 |
|
|
Reflux gastro-oesophagien |
|
26 (2,4) |
|
1 (< 0,1) |
|
|
Hémorragie rectale |
|
14 (1,3) |
|
4 (0,4) |
|
|
Sécheresse de la bouche |
|
19 (1,7) |
|
2 (0,2) |
|
|
Distension abdominale |
|
14 (1,3) |
|
1 (< 0,1) |
|
|
Stomatite |
|
46 (4,2) |
|
2 (0,2) |
|
|
|
Iléus* |
|
|
7 (0,6) |
5 (0,5) |
|
|
Gastrite |
|
|
10 (0,9) |
0 |
|
|
Colite* |
|
|
10 (0,9) |
5 (0,5) |
|
|
Perforation gastro-intestinale |
|
|
3 (0,3) |
1 (<0,1) |
|
|
Hémorragie gastro-intestinale |
|
|
2 (0,2) |
1 (<0,1) |
|
Affection de la peau et du tissu sous-cutané |
Alopécie |
|
80 (7,3) |
|
0 |
|
Sécheresse de la peau |
|
23 (2,1) |
|
0 |
|
|
Erythème |
|
|
8 (0,7) |
0 |
|
|
Altération des ongles |
|
18 (1,6) |
|
|
|
|
Affections musculo-squelettiques et systémiques |
Douleur dorsale |
166 (15,2) |
|
|
24 (2,2) |
|
Arthralgie |
|
88 (8,1) |
|
9 (0,8) |
|
|
Douleur des extrêmités |
|
76 (7,0) |
|
9 (0,8) |
|
|
Spasmes musculaires |
|
51 (4,7) |
|
0 |
|
|
Myalgie |
|
40 (3,7) |
|
2 (0,2) |
|
|
Douleur thoracique musculo-squelettique |
|
34 (3,1) |
|
3 (0,3) |
|
|
Faiblesse musculaire |
|
31 (2,8) |
|
1 (0,2) |
|
|
Douleur au niveau du flanc |
|
17 (1,6) |
|
5 (0,5) |
|
|
Affections du rein et des voies urinaires |
Insuffisance rénale aigue |
|
21 (1,9) |
|
14 (1,3) |
|
Insuffisance rénale |
|
|
8 (0,7) |
6 (0,5) |
|
|
Dysurie |
|
52 (4,8) |
|
0 |
|
|
Colique rénale |
|
14 (1,3) |
|
2 (0,2) |
|
|
Hématurie |
205 (18,8) |
|
|
33 (3,0) |
|
|
Pollakiurie |
|
26 (2,4) |
|
2 (0,2) |
|
|
Hydronéphrose |
|
25 (2,3) |
|
13 (1,2) |
|
|
Rétention urinaire |
|
36 (3,3) |
|
4 (0,4) |
|
|
Incontinence urinaire |
|
22 (2,0) |
|
0 |
|
|
Obstruction urétrale |
|
|
8 (0,7) |
6 (0,5) |
|
|
Affections des organes reproducteurs et du sein |
Douleur pelvienne |
|
20 (1,8) |
|
5 (0,5) |
|
Troubles généraux et anomalies au site d’administration |
Fatigue |
333 (30,5) |
|
|
42 (3,8) |
|
Asthénie |
227 (20,8) |
|
|
32 (2,9) |
|
|
Fièvre |
|
90 (8,2) |
|
5 (0,5) |
|
|
Oedème périphérique |
|
96 (8,8) |
|
2 (0,2) |
|
|
Inflammation des muqueuses |
|
23 (2,1) |
|
1 (<0,1) |
|
|
Douleur |
|
36 (3,3) |
|
7 (0,6) |
|
|
Douleur thoracique |
|
11 (1,0) |
|
2 (0,2) |
|
|
Oedème |
|
|
8 (0,7) |
1 (<0,1) |
|
|
Frissons |
|
12 (1,1) |
|
0 |
|
|
Malaise |
|
21 (1,9) |
|
0 |
|
|
Investigations |
Perte de poids |
|
81 (7,4) |
|
0 |
|
Augmentation de l’Aspartate aminotransferase |
|
13 (1,2) |
|
1 (<0,1) |
|
|
Augmentation des Transaminases |
|
|
7 (0,6) |
1 (<0,1) |
|
a basé sur les valeurs du laboratoire
* voir rubriques détaillées ci-dessous
Description de certains effets indésirables
Neutropénies et évènements cliniques associés
L’utilisation du G-CSF a montré qu’elle limitait l’incidence et la sévérité des neutropénies (voir rubriques 4.2 et 4.4).
L’incidence des neutropénies de grade ≥ 3, basée sur les données de laboratoire variait de 44,7% à 76,7% en fonction de l’utilisation de G-CSF, l’incidence la plus faible étant rapportée lorsque la prophylaxie par G-CSF était utilisée. De même, l’incidence des neutropénies fébriles de grade ≥3 variait entre 3,2% et 8,6%.
Les complications de neutropéniques (incluant des neutropénies fébriles, des infections neutropéniques/état septique, et des neutropénies coliques) qui, dans certains cas, ont entrainé une issue fatale, ont été rapportées chez 4,0% des patients lorsqu’une prophylaxie primaire par G-CSF était utilisée, et chez 12,8% des patients dans le cas contraire.
Troubles cardiaques et arythmies
Dans l’analyse des données combinées, les évènements cardiaques, , étaient rapportés chez 5,5% des patients dont 1,1% ont présentés des arythmies cardiaques de grade ≥ 3. L’incidence de la tachycardie dans le bras cabazitaxel était de 1,0%, dont moins de 0,1% était de grade ≥ 3. L’incidence de la fibrillation auriculaire était de 1,3%. Des cas d’insuffisance cardiaque ont été rapportés pour 2 patients (0,2%), dont un a été d’issue fatale. Une fibrillation ventriculaire fatale a été rapportée chez 1 patient (0,3%), et un arrêt cardiaque chez 3 patients (0,5%). Aucun de ces évènements n’a été considéré comme relié par les investigateurs.
Hématuries
Dans l’analyse des données combinées, la fréquence des hématuries, quels que soient leurs grades, étaient de 18,8% à la dose de 25 mg/m2 (voir rubrique 5.1). Lorsque cela était documenté, des facteurs de confusion tels que la progression de la maladie, instrumentation, infection ou traitement par anticoagulant/AINS/ acide acétylsalicylique ont été identifiés dans près de la moitié des cas.
Autres anomalies biologiques
Dans l’analyse poolée, l’incidence des anémies de grade ≥ 3, des augmentations des ASAT, ALAT, et bilirubine basées sur des examens biologiques étaient respectivement de 12,0%, 1,3%, 1,0% et 0,5%.
Affections gastro-intestinales
Des colites (comprenant des entérocolites et des entérocolites neutropéniques), et des gastrites ont été observées. Des hémorragies gastro-intestinales, des perforations gastro-intestinales, et des iléus (obstructions intestinales) ont également été rapportés (voir rubrique 4.4).
Affections respiratoires
Des cas de pneumonies interstitielles/pneumopathies inflammatoires et de pneumopathies interstitielles diffuses, parfois fatals ont été rapportés avec une fréquence inconnue (ne peut être estimée à partir des données disponibles).
Affections rénales et urinaires
Des cystites dues à un phénomène de rappel après radiothérapie, incluant des cystites hémorragiques, ont été peu fréquemment rapportées.
Population pédiatrique
Voir la rubrique 4.2
Autres populations particulières
Sujets âgés
Sur les 1092 patients traités par cabazitaxel à la dose de 25 mg/m2 dans les études sur le cancer de la prostate, 755 patients étaient âgés de 65 et plus, incluant 238 patients de plus de 75 ans.
Les effets indésirables non hématologiques suivants étaient rapportés avec un taux ≥ 5%, chez des patients de plus de 65 ans, comparés aux patients plus jeunes : fatigue (33,5% versus 23,7 %), asthénie (23,7% versus 14,2%), constipation (20,4% versus 14,2%) et dyspnée (10,3% versus 5,6%).
La neutropénie (90,9% versus 81,2%) et la thrombocytopénie (48,8% versus 36.1%) étaient également 5% plus élevés chez les patients de 65 ans ou plus par rapport aux patients plus jeunes. La neutropénie de grade ≥3 et la neutropénie fébrile étaient rapportées avec la plus grande différence de taux entre les deux groupes d’âge (respectivement 14% et 4% plus élevée chez les patients ≥ 65 ans comparé aux patients < 65 ans) (voir rubriques 4.2 et 4.4) .
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Il n’y pas d’antidote au cabazitaxel. Les complications prévisibles liées au surdosage consisteraient en une exacerbation des effets indésirables, tels qu’une aplasie médullaire et des troubles gastro-intestinaux.
En cas de surdosage, le patient doit être admis dans une unité spécialisée afin de surveiller étroitement ses fonctions vitales. Les patients doivent recevoir un traitement par G-CSF dès que possible après découverte du surdosage. D’autres mesures symptomatiques appropriées doivent être prises.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Agents antinéoplasiques, Code ATC : L01CD04.
Mécanisme d’action
Le cabazitaxel est un agent antinéoplasique qui agit en perturbant le réseau de microtubules dans les cellules. Le cabazitaxel se lie à la tubuline et favorise l’assemblage de la tubuline en microtubules, tout en inhibant leur dépolymérisation. Ceci conduit à la stabilisation des microtubules, ce qui entraîne l’inhibition de la mitose et l’interphase des fonctions cellulaires.
Effets pharmacodynamiques
Le cabazitaxel a fait preuve d’un large spectre d’activité antitumorale contre des tumeurs humaines au stade avancée greffées chez la souris. Le cabazitaxel est actif sur les tumeurs sensibles au docétaxel. En outre, le cabazitaxel a démontré une activité dans des modèles tumoraux insensibles à la chimiothérapie, y compris le docétaxel.
Efficacité et sécurité clinique
L’efficacité et la tolérance de cabazitaxel en association à la prednisone ou la prednisolone, ont été évaluées dans une étude de phase III randomisée, ouverte, internationale, multicentrique (étude EFC6193), chez des patients avec un cancer de la prostate métastatique résistant à la castration, précédemment traités par un traitement à base de docétaxel.
La Survie Globale (SG) a été le critère principal d’efficacité de l’étude.
Les critères secondaires étaient la Survie Sans Progression [SSP (définie comme le temps entre la randomisation et la progression tumorale), la progression de l’antigène prostatique spécifique PSA, la progression de la douleur ou le décès quelle qu’en soit la cause, selon l’évènement d’origine], le taux de réponse tumorale basé sur la réponse aux critères d’ évaluation des tumeurs solides (RECIST), la Progression du PSA (défini comme Non Répondeurs si ≥ 25% d’augmentation ou comme Répondeurs si >50% du PSA), réponse du PSA (baisse des taux de PSA sériques au moins de 50%), progression de la douleur [évaluée en utilisant l’échelle d’intensité de la douleur présente (PPI) issue du questionnaire McGill-Melzack et du score Analgésique (AS)] et de la réponse à la douleur (définie comme une réduction de plus de 2 points à partir de la baseline médiane du PPI sans augmentation concomitante de l’AS, ou une diminution de ≥ 50% d’utilisation d’analgésique à partir de la baseline moyenne de l’AS sans augmentation concomitante de la douleur).
Un total de 755 patients ont été randomisés pour recevoir soit cabazitaxel 25mg/m2 par voie intraveineuse, toutes les 3 semaines, pendant un maximum de 10 cycles avec de la prednisone ou prednisolone 10 mg par jour par voie orale (n=378), soit mitoxantrone 12 mg/m2 par voie intraveineuse, toutes les 3 semaines avec un maximum de 10 cycles avec de la prednisone ou prednisolone 10 mg par jour par voie orale (n=377).
Dans cette étude ont été inclus des patients âgés de plus de 18 ans ayant un cancer de la prostate métastatique résistant à la castration soit avec maladie mesurable selon les critères RECIST soit non-mesurable avec une élévation du niveau de PSA ou apparition de nouvelles lésions et un état général (statut de performance) de 0 à 2 selon l’Eastern Cooperative Oncology Group (ECOG). Les patients devaient avoir un taux de neutrophiles > 1 500/mm3, un taux de plaquettes > 100 000/mm3, un taux d’hémoglobine > 10 g/dl, une créatinine < 1.5 x LSN, une bilirubine totale < 1 x la -Limite Supérieure de la Normale (LSN), ASAT et ALAT < 1.5 x LSN.
Les patients avec des antécédents d’insuffisance cardiaque congestive ou d’infarctus du myocarde dans les 6 derniers mois ou les patients avec des arythmies cardiaques non contrôlées, une angine de poitrine et/ou une hypertension , n’ont pas été inclus dans cette étude.
Les données démographiques, incluant l’âge, ethnie et état général selon l’ECOG (0 à 2) étaient équilibrés dans les bras de traitement. Dans le groupe cabazitaxel, la moyenne d’âge était 68 ans, écart (46-92) et la répartition ethnique était 83,9 % Caucasien, 6,9% Asiatique/Oriental, 5,3 % Noir et 4% Autres.
Le nombre de cycles médian était de 6 dans le groupe cabazitaxel et de 4 dans le groupe mitoxantrone. Le nombre de patients ayant complété le traitement à l’étude (10 cycles) était respectivement 29,4 % dans le groupe cabazitaxel et 13,5 % dans le groupe comparateur.
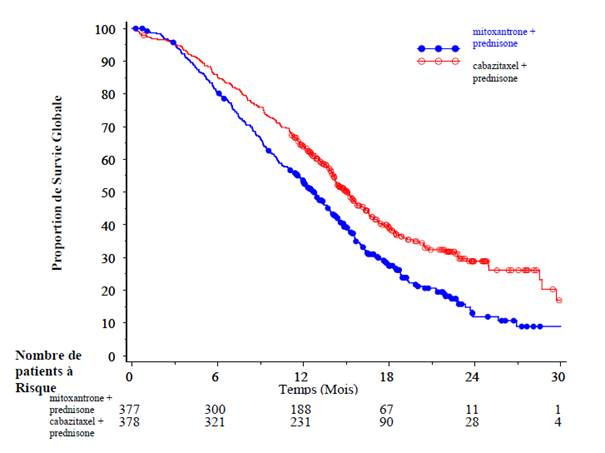
La Survie Globale était significativement plus longue avec cabazitaxel comparée à mitoxantrone (15,1 mois versus 12,7 mois respectivement), avec une réduction du risque de décès de 30 % comparée à la mitoxantrone (voir tableau 3 et figure 1).
Un sous-groupe de 59 patients a préalablement reçu une dose cumulative de docétaxel <225 mg / m² (29 patients dans le bras cabazitaxel, 30 patients dans le bras mitoxantrone. Il n'y avait pas de différence significative de la survie globale dans ce groupe de patients (HR (95% CI) 0,96 (0,49 1,86)).
Tableau 3 – Efficacité de cabazitaxel dans l’étude EFC6193 dans le traitement des patients ayant un cancer de la prostate métastatique résistant à la castration
|
|
cabazitaxel + prednisone n=378 |
mitoxantrone + prednisone n=377 |
|
Survie Globale |
|
|
|
Nombre de patients décédés (%) |
234 (61,9%) |
279 (74%) |
|
Survie médiane (mois) (IC 95%) |
15,1 (14,1-16,3) |
12.7 (11,6-13,7) |
|
Hazard Ratio (HR)1 (IC 95%) |
0,70 (0,59-0,83) |
|
|
Valeur de p |
<0,0001 |
|
1HR estimé selon le modèle de Cox ; un hazard ratio inférieur à 1 est en faveur de cabazitaxel
Figure 1 : Courbes de Survie Globale de Kaplan Meier (EFC6193)
Il y a eu une amélioration de la SSP dans le bras cabazitaxel comparé au bras mitoxantrone : respectivement 2,8 mois (2,4 -3,0) versus 1,4 mois (1,4 - 1,7), HR (95% IC) 0,74 (0,64 - 0,86), p<0,0001.
Il y a eu un taux de réponse tumorale significativement plus élevé de 14,4% (95% IC : 9,6 – 19,3) chez les patients dans le bras cabazitaxel comparé au 4,4% (95% IC : 1,6 – 7,2) chez les patients dans le bras mitoxantrone, p=0,0005.
Les critères secondaires PSA étaient positifs dans le bras cabazitaxel. Il y avait une progression médiane du PSA de 6,4 mois (95% IC : 5,1 – 7,3) pour les patients dans le bras cabazitaxel, comparé aux 3.1 mois (95% IC : 2,2 – 4,4) dans le bras mitoxantrone, HR 0.75 mois (95% IC : 0,63 – 0,90), p=0,0010. La Réponse du PSA était de 39,2% chez les patients dans le bras cabazitaxel (95% IC : 33,9 – 44,5) versus 17,8% des patients sous mitoxantrone (95% IC : 13,7 – 22,0), p=0,0002.
Il n’y a pas eu de différence statistique entre les deux bras de traitement concernant la progression de la douleur et la réponse à la douleur.
Dans une étude de phase III, de non-infériorité, multicentrique, internationale, randomisée, en ouvert (étude EFC11785), 1200 patients avec un cancer de la prostate métastatique résistant à la castration, précédemment traités par un traitement à base de docétaxel, ont été randomisés pour recevoir du cabazitaxel soit à la dose de 25 mg/m2 (n = 602) soit à la dose de 20 mg/m2 (n = 598). La Survie Globale (SG) était le critère principal d’efficacité.
L’étude a atteint son objectif principal en démontrant la non-infériorité du cabazitaxel à la dose de 20 mg/m2 comparée à la dose de 25 mg/m2 (voir tableau 4). Un pourcentage significativement plus élevé (p<0,001) de patients a montré une réponse du PSA dans le groupe 25 mg/m2 (42,9 %) par rapport au groupe 20 mg/m2 (29,5 %). Un risque de progression du PSA significativement plus élevé a été observé chez les patients traités à la dose de 20 mg/m2 par rapport à la dose de 25 mg/m2 (HR 1,195 ; IC 95 % : 1,025 à 1,393). Il n’y a pas eu de différence statistiquement significative en ce qui concerne les autres critères secondaires (SSP, réponse tumorale et réponse à la douleur, progression de la tumeur et progression de la douleur et quatre sous-catégories du questionnaire FACT-P (Functional Assessment of Cancer Therapy-Prostate)).
Tableau 4 – Survie globale dans l’étude EFC11785 dans le bras cabazitaxel 25 mg/m2 versus bras cabazitaxel 20 mg/m2 (Analyse en intention de traiter) – Critère principal d’efficacité
|
|
CBZ20+PRED n=598 |
CBZ25+PRED n=602 |
|
Survie Globale |
|
|
|
Nombre de décès, n (%) |
497 (83,1%) |
501 (83,2%) |
|
Survie médiane (IC 95%) (mois) |
1,4 (12,19 to 1,88) |
14.5 (13,47 to 15,28) |
|
Hazard Ratioa |
|
|
|
versus CBZ25+PRED |
1,024 |
- |
|
ICS unilatérale 98,89% |
1,184 |
- |
|
ICI unilatérale 95% |
0,922 |
- |
CBZ20 = Cabazitaxel 20 mg/m2, CBZ25 = Cabazitaxel 25 mg/ m2, PRED = Prednisone/Prednisolone
IC = intervalle de confiance, ICI = limite inférieure de l’intervalle de confiance, ICS = limite supérieure de l’intervalle de confiance
a Le Hazard Ratio est estimé à l’aide d’un modèle de régression de Cox, à risque proportionnel. Un hazard ratio < 1 indique un risque plus faible avec le cabazitaxel à la dose de 20 mg/m2 par rapport à la dose de 25 mg/m2.
Le profil de tolérance du cabazitaxel 25 mg/m2 dans l’étude EFC11785 était qualitativement et quantitativement similaire à celui observé dans l’étude EFC6193. L’étude EFC11785 a mis en évidence un meilleur profil de tolérance avec le cabazitaxel à la dose de 20 mg/m².
Tableau 5 – Résumé des données de tolérance dans le bras cabazitaxel 25 mg/m² versus bras cabazitaxel 20 mg/m² dans l’étude EFC11785
|
|
CBZ20+PRED n=580 |
CBZ25+PRED n=595 |
|
Nombre médian de cycles/ durée médiane de traitement |
6/ 18 semaines |
7/ 21 semaines |
|
Nombre de patients avec réduction de dose n (%)
|
de 20 à 15 mg/m2: 58 (10,0%) de 15 à 12 mg/m2: 9 (1,6%) |
de 25 à 20 mg/m2: 128 (21,5%) de 20 à 15 mg/m2: 19 (3,2%) de 15 à 12 mg/m2: 1 (0,2%) |
|
Effets indésirables tous gradesa (%) |
||
|
Diarrhées |
30,7 |
39,8 |
|
Nausées |
24,5 |
32,1 |
|
Fatigue |
24,7 |
27,1 |
|
Hématurie |
14,1 |
20,8 |
|
Asthénie |
15,3 |
19,7 |
|
Diminution de l’appétit |
13,1 |
18,5 |
|
Vomissements |
14,5 |
18,2 |
|
Constipation |
17,6 |
18,0 |
|
Douleur dorsale |
11,0 |
13,9 |
|
Neutropénie clinique Neutropénie |
3,1 |
10,9 |
|
Infection du tractus urinaire |
6,9 |
10,8 |
|
Neuropathie périphérique sensitive |
6,6 |
10,6 |
|
Dysgueusie |
7,1 |
10,6 |
|
Effets indésirables de grade ≥ 3b (%) |
||
|
Neutropénie clinique |
2,4 |
9,6 |
|
Neutropénie febrile
|
2,1 |
9,2 |
|
Anomalies hématologiquesc (%) |
||
|
Neutropénie de grade ≥ 3 |
41,8 |
73,3 |
|
Anémie de grade ≥ 3 |
9,9 |
13,7 |
|
Thrombopénie de grade ≥ 3 |
2,6 |
4,2 |
CBZ20 = Cabazitaxel 20 mg/m2, CBZ25 = Cabazitaxel 25 mg/ m2, PRED = Prednisone/Prednisolone
a Effets indésirables tous grades confondus avec une incidence supérieure à 10%
b Effets indésirables de grade ≥ 3 avec une incidence supérieure à 5%
c Basés sur des valeurs de laboratoire
Dans une étude de phase IV prospective, multinationale, randomisée, avec un contrôle actif, en ouvert, (LPS14201/étude CARD), 255 patients atteints d’un cancer de la prostate métastatique résistant à la castration (CPRCm) précédemment traités, quel que soit l’ordre, par un traitement à base de docétaxel et des agents ciblant les récepteurs androgéniques (ARTA) (abiratérone or enzalutamide, avec une progression dans les 12 mois suivant l’initiation du traitement), ont été randomisés pour recevoir soit cabazitaxel 25 mg/m2 toutes les 3 semaines plus prednisone/prednisolone 10 mg par jour (n=129) ou ARTA (abiratérone 1000 mg une fois par jour plus prednisone/prednisolone 5 mg deux fois par jour ou enzalutamide 160 mg une fois par jour) (n=126). La survie sans progression radiographique (SSPr) définie par le Prostate Cancer Working Group-2 (PCWG2) était le critère principal d’évaluation. Les critères secondaires incluaient la survie globale, la survie sans progression, la réponse du PSA et la réponse tumorale.
Les caractéristiques démographiques et de la pathologie étaient équilibrées entre les 2 bras de traitement. A l’inclusion, l’âge médian global était de 70 ans, 95% des patients avaient un score de performance ECOG entre 0 et 1 et le score de Gleason médian était de 8. Soixante et un pour cent (61%) des patients avaient reçu l’ARTA après le docétaxel.
L’étude a atteint le critère principal d’évaluation : la SSPr était significativement plus longue avec cabazitaxel comparée à l’ARTA (8,0 mois versus 3,7 respectivement), avec une réduction du risque de progression radiographique de 46% comparée à l’ARTA (voir tableau 6 et figure 2).
Tableau 6 - Efficacité de cabazitaxel dans l’étude CARD dans le traitement des patients atteints de cancer de la prostate résistant à la castration (analyse en intention de traiter) – Survie sans progression radiographique (SSPr)
|
|
Cabazitaxel + prednisone/prednisolone + G-CSF
n=129 |
ART : Abiratérone + prednisone/prednisolone ou Enzalutamide n=126 |
|
Nombre d’évènements à la date butoir (%) |
95 (73,6%) |
101 (80,2%) |
|
SSPr médian (mois) (95% CI) |
8,0 (5,7 à 9,2) |
3,7 (2,8 à 5,1) |
|
Hazard ratio (HR) (95% CI) |
0,54 (0,40 to 0,73) |
|
|
Valeur de p1 |
< 0,0001 |
|
1test log-rank stratifié, seuil de significativité = 0,05
Figure 2 – Critère principal - Graphe Kaplan-Meier de la SSP radiographique (population ITT)
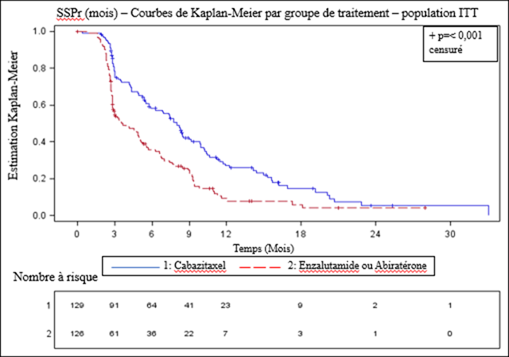
Les marques indiquent les données censurées.
Les analyses planifiées en sous-groupes de la SSPr basées sur des facteurs de stratification à la randomisation ont produit un hazard ratio de 0,61 (IC à 95% : 0,39 to 0,96) chez les patients qui ont reçu l’ARTA antérieur avant le docétaxel et un hazard ratio de 0,48 (IC à 95% : 0,32 to 0,70) chez les patients qui ont reçu l’ARTA antérieur après le docétaxel.
Le cabazitaxel était statistiquement supérieur aux comparateurs ARTA pour chaque critère secondaire clé protégé par alpha, incluant la survie globale (13,6 mois pour le bras cabazitaxel versus 11,0 mois pour le bras ARTA, HR 0,64, IC à 95%: 0,46 to 0,89; p=0,008), la survie sans progression (4,4 mois pour le bras cabazitaxel versus 2,7 mois pour le bras ARTA, HR 0,52; IC à 95%: 0,40 to 0,68), la réponse confirmée du PSA (36,3% pour le bras cabazitaxel versus 14,3% pour le bras ARTA, p=0,0003) et la meilleure réponse tumorale (36,5% pour le bras cabazitaxel versus 11,5% pour le bras ARTA, p=0,004).
Le profil de tolérance de cabazitaxel 25 mg/m2 observé dans l’étude card était globalement cohérent avec celui observé dans les études TROPIC et PROSELICA (voir rubrique 4.8). L’incidence d’effets indésirables de grade ≥ 3 était 53,2% dans le bras cabazitaxel arm versus 46,0% dans le bras ARTA. L’incidence d’effets indésirables graves de grade ≥ 3 était 31,7% dans le bras cabazitaxel versus 37,1% dans le bras arta.
L’incidence de patients ayant arrêté définitivement le traitement à l’étude en raison d’effets indésirables était 19,8% dans le bras cabazitaxel versus 8,1% dans le bras ARTA. L’incidence de patients ayant un effet indésirable fatal était 5,6% dans le bras cabazitaxel versus 10,5% dans le bras ARTA.
Population pédiatrique
L'Agence européenne des médicaments a renoncé à l'obligation de soumettre les résultats des études du produit de référence contenant du cabazitaxel dans tous les sous-ensembles de la population pédiatrique dans l'indication du cancer de la prostate (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
Cabazitaxel a été évalué dans une étude ouverte, multicentrique de phase 1/2 conduite chez 39 patients pédiatriques (âgés de 4 à 18 ans pour la phase 1 et de 3 à 16 ans pour la phase 2). La phase 2 n’a pas démontré l’efficacité du cabazitaxel administré en monothérapie dans la population pédiatrique présentant des gliomes infiltrants du tronc cérébral (GITC) récidivants ou réfractaires et des gliomes de haut grade (GHG) traités à la dose de 30 mg/m².
5.2. Propriétés pharmacocinétiques
Absorption
Après une administration intraveineuse d’une heure de cabazitaxel à 25mg/m² chez des patients avec un cancer de la prostate métastatique (n=67), la Cmax était de 226 ng/ml (Coefficient de Variation (CV) : 107%) et était atteint à la fin d’une perfusion de 1 heure (Tmax). L’AUC moyenne (aire sous la courbe) était de 991 ng.h/ml (CV : 34%). Pas de déviation majeure proportionnelle à la dose n’a été observée de 10 à 30 mg/m² chez des patients avec des tumeurs solides avancées (n=126).
Distribution
Le volume de distribution (VSS) était de 4870 L (2640 L/m² pour un patient avec une surface corporelle médiane de 1,84 m²) à l’état d’équilibre.
In vitro, la liaison du cabazitaxel aux protéines sériques humaines était de 89-92% et n’était pas saturable jusqu’à 50 000 ng/ml, ce qui couvre les concentrations maximales observées dans les études cliniques. Le cabazitaxel est principalement lié à l’albumine sérique humaine (82,0%) et aux lipoprotéines (87,9% pour HDL, 69,8% pour LDL, et 55,8% pour VLDL). Le rapport des concentrations in vitro sang-plasma dans le sang humain varie entre 0,90 et 0,99, indiquant que le cabazitaxel était distribué de façon égale entre le sang et le plasma.
Biotransformation
Le cabazitaxel est largement métabolisé dans le foie (>95%), principalement par l’isoenzyme CYP3A (80 à 90%). Le cabazitaxel est le principal composé circulant dans le plasma humain. Sept métabolites ont été détectés dans le plasma (incluant 3 métabolites actifs issus de la o-déméthylation), avec le principal représentant 5% de l’exposition de la molécule mère administrée. Environ 20 métabolites du cabazitaxel sont excrétés dans les urines et fèces humaines.
A partir des études in vitro, le risque potentiel d’inhibition avec le cabazitaxel à des concentrations cliniquement pertinentes est possible vis à vis des produits médicamenteux qui sont principalement des substrats du CYP3A.
Cependant une étude clinique a démontré que cabazitaxel (25 mg/m2, administré en perfusion unique d’une heure) ne modifie pas les taux plasmatiques de midazolam, un substrat-test du CYP3A. Par conséquent, aux doses thérapeutiques, la co-administration de substrats du CYP3A et de cabazitaxel ne devrait pas avoir d’impact clinique.
Il n’y a pas de risque potentiel d’inhibition des produits médicamenteux qui sont des substrats d’autres enzymes du CYP (1A2, 2B6, 2C9, 2C8, 2C19, 2E1 et 2D6) de même qu’il n’y a pas de risque potentiel d’induction par le cabazitaxel sur des produits médicamenteux substrats du CYP1A, CYP2C9 et CYP3A. Le cabazitaxel n’a pas inhibé in vitro la principale voie de biotransformation de la warfarine en 7-hydroxywarfarine qui est médiée par le CYP2C9. Par conséquent, aucune interaction pharmacocinétique du cabazitaxel sur la warfarine n’est attendue in vivo.
In vitro le cabazitaxel n’a pas inhibé les Protéines Multidrogues Résistantes (MRP) : MRP1 et MRP2 ou le transporteur de cations organiques (OCT1). Cabazitaxel inhibe le transport assuré par la P-glycoprotéine (PgP) (digoxine, vinblastine), par les Protéines Résistantes au Cancer du Sein (BCRP) (méthotrexate) et par le polypeptide transporteur d’anions organiques OATP1B3 (CCK8) à des concentrations au moins 15 fois supérieures à ce qui est observé en clinique tandis qu’il inhibe le transport par OATP1B1 (estradiol-17β-glucuronide) à des concentrations seulement 5 fois supérieures à ce qui est observé en clinique. Par conséquent, le risque d’interaction avec les substrats des MRP, de l’OCT1, de la PgP, de la BRCP et de l’OATP1B3 est peu probable in vivo à la dose de 25 mg/m². Le risque d’interaction avec le transporteur OATP1B1 existe notamment pendant la durée de la perfusion (1 heure) et jusqu’à 20 minutes après la fin de la perfusion (voir rubrique 4.5).
Élimination
Après 1 heure de perfusion intraveineuse de (14C)-cabazitaxel à 25mg/m², environ 80% de la dose administrée étaient éliminée en moins de 2 semaines. Le cabazitaxel est principalement éliminé dans les fèces en nombreux métabolites (76% de la dose); tandis que l’excrétion rénale du cabazitaxel et de ses métabolites représente moins de 4% de la dose (2,3% comme produit inchangé dans les urines).
Le cabazitaxel avait une forte clairance plasmatique de 48.5 l/h (26,4 l/h/m² pour un patient avec une surface corporelle moyenne de 1,84 m²) et une demi-vie longue de 95 heures.
Populations particulières
Patients âgés
Dans les analyses pharmacocinétiques de population chez 70 patients de 65 ans et plus (57 de 65 à 75 ans et 13 patients de plus de 75 ans), aucun effet âge sur les paramètres pharmacocinétiques du cabazitaxel n’a été observé.
Patients pédiatriques
La tolérance et l’efficacité de cabazitaxel n’ont pas été encore établies chez l’enfant et chez les adolescents de moins de 18 ans.
Insuffisants hépatiques
Le cabazitaxel est éliminé principalement par métabolisation hépatique.
Une étude spécifique chez 43 patients ayant un cancer et une insuffisance hépatique a montré l’absence d’influence de l’insuffisance hépatique légère (bilirubine totale > 1 à ≤ 1,5 x LSN ou ASAT > 1,5 x LSN) sur la pharmacocinétique du cabazitaxel. La Dose Maximale Tolérée (DMT) du cabazitaxel était 20 mg/m2.
Chez 3 patients ayant une insuffisance hépatique sévère (bilirubine totale > 3 x LSN), une baisse de 39 % de la clairance a été observée comparativement aux patients ayant une insuffisance hépatique légère, montrant l’effet de l’insuffisance hépatique sévère sur la pharmacocinétique du cabazitaxel. La DMT du cabazitaxel chez les patients ayant une insuffisance hépatique sévère n’a pas été établie.
Basée sur la tolérance et les données de sécurité, la dose du cabazitaxel doit être réduite chez les patients ayant une insuffisance hépatique légère (voir rubriques 4.2, 4.4). Cabazitaxel est contre-indiqué chez les patients ayant une insuffisance hépatique modérée et sévère (voir rubrique 4.3).
Insuffisants rénaux
Le cabazitaxel est à peine excrété par les reins (2,3% de la dose). Une analyse pharmacocinétique de population réalisée chez 170 patients dont 14 patients inclus avec une insuffisance rénale modérée (clairance de la créatinine comprise entre 30 et 50 ml/min) et 59 patients avec une insuffisance rénale légère (clairance de la créatinine comprise entre 50 et 80 ml/min), a montré qu’une insuffisance rénale légère à modérée n’a pas d’effet significatif sur la pharmacocinétique du cabazitaxel. Cela a été confirmé par une étude pharmacocinétique comparative chez des patients atteints d’une tumeur solide avec une fonction rénale normale (8 patients), une insuffisance rénale modérée (8 patients) et sévère (9 patients), qui ont reçu plusieurs cycles de cabazitaxel en perfusion jusqu’à 25 mg/m2.
5.3. Données de sécurité préclinique
Des réactions indésirables non observées dans les études cliniques, mais vues chez le rat lors d’études de toxicité à doses répétées, à des niveaux d’exposition plus élevés que les niveaux d’exposition clinique et pouvant être pertinents en clinique, étaient des troubles oculaires caractérisés par un gonflement/dégénération de la fibre optique sous-capsulaire. Ces effets étaient partiellement réversibles après 8 semaines.
Aucune étude de carcinogénicité n’a été menée avec le cabazitaxel.
Cabazitaxel n’a pas induit de mutations dans le test de mutation reverse bactérienne (Ames). Il n’était pas clastogénique dans les tests in-vitro dans les lymphocytes humains (pas d’induction d’aberration structurale chromosomique mais il augmentait le nombre de cellules polyploïdes) et a induit une augmentation des micronoyaux dans les tests in-vivo chez les rats. Cependant ces résultats de génotoxicité sont inhérents à l’activité pharmacologique de la molécule (inhibition de la dépolymérisation de la tubuline) et ont été observés avec des produits médicamenteux présentant la même activité pharmacologique.
Le cabazitaxel n’a pas d’incidence sur les performances d’accouplement ou la fertilité des rats males traités. Cependant, dans les études de toxicité à doses-répétées, une dégénération de la vésicule séminale et une atrophie du tubule séminifère dans les testicules ont été observés chez le rat et une dégénération testiculaire (nécrose minime des cellules épithéliales uniques dans l’épididyme) a été observée chez les chiens. Les expositions chez les animaux étaient similaires ou plus faibles que celles vues chez les humains recevant des doses cliniquement pertinentes de cabazitaxel.
Le cabazitaxel a induit une toxicité embryofoetale chez le rat femelle traité par voie intraveineuse une fois par jour depuis le 6ème jour gestationnel au 17ème, liée à une toxicité maternelle et consistant à des morts foetales et une diminution du poids moyen foetal associée à un retard dans l’ossification du squelette. Les expositions chez l’animal étaient plus basses que celles vues chez les humains recevant des doses cliniquement pertinentes de cabazitaxel. Le cabazitaxel passe la barrière placentaire chez le rat.
Chez les rats, le cabazitaxel et ses métabolites sont excrétés dans le lait maternel à une quantité pouvant aller jusqu’à 1,5% de la dose administrée sur 24 heures.
Évaluation du risque environnemental
Les résultats des études d’évaluation du risque environnemental ont indiqué que l’utilisation de cabazitaxel n’aura pas de risque significatif sur l’environnement aquatique (voir rubrique 6.6 élimination des médicaments non utilisés).
Polysorbate 80
Acide citrique
Solvant :
Ethanol 96%
Eau pour préparations injectables.
Les poches de perfusion en PVC ou les sets de perfusion en polyuréthane ne doivent pas être utilisées pour la préparation et l’administration de la solution pour perfusion.
Flacons non ouverts
3 ans
Après ouverture
Les flacons de solution et de solvant doivent être utilisés immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l’utilisateur.
Après la dilution initiale de la solution avec le solvant
La stabilité physico-chimique a été démontrée pendant 1 heure à température ambiante (15°C - 30°C) et de 21 jours à 2°C-8°C.
D’un point de vue microbiologique, le mélange solution à diluer et solvant doit être utilisé immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l'utilisateur et ne devraient pas normalement dépasser 24 heures à 2°C - 8°C, sauf en cas de dilution réalisée dans des conditions d’asepsie contrôlées et validées.
Après la dilution finale dans la poche de perfusion/le flacon
La stabilité physico-chimique de la solution pour perfusion a été démontrée pendant 8 heures à température ambiante (15°C-30°C) (incluant l’heure d’administration de la perfusion) et pendant 48 heures au réfrigérateur (2°C-8°C) (incluant l’heure d’administration de la perfusion) dans la poche de perfusion et la stabilité physico-chimique de la solution pour perfusion a été démontrée pendant 8 heures à température ambiante (15°C-30°C) (incluant l’heure d’administration de la perfusion) dans le flacon en verre.
D’un point de vue microbiologique, la solution pour perfusion doit être utilisée immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l’utilisateur et ne devraient pas normalement dépasser 24 heures à 2°C - 8°C, sauf en cas de dilution réalisée dans des conditions d’asepsie contrôlées et validées.
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
Une boîte contient 1 flacon de solution à diluer et 1 flacon de solvant.
· Solution à diluer : 1,5 ml de solution à diluer, en flacon en verre transparent (type I) de 15 ml fermé par un bouchon en caoutchouc scellé par une capsule de type flip-off. Chaque flacon contient 60 mg de cabazitaxel pour 1,5 ml en volume nominal (volume de remplissage : 73,2 mg de cabazitaxel/ 1,83 ml). Ce volume de remplissage a été établi pendant le développement de CABAZITAXEL REDDY PHARMA afin de compenser la perte de liquide survenant au cours de la préparation du prémélange. Ce suremplissage permet après dilution avec la totalité du solvant de CABAZITAXEL REDDY PHARMA d’obtenir le volume minimal de prémélange extractible de 6 ml, contenant 10 mg/ml de CABAZITAXEL REDDY PHARMA, ce qui correspond à la quantité figurant sur l’étiquette, à savoir 60 mg par flacon.
· Solvant : 4,5 ml de solvant en flacon en verre transparent (type I) de 15 ml fermé par un bouchon en caoutchuc scellé par une capsule de type flip-off. Chaque flacon contient 4,5 ml en volume nominal (volume de remplissage : 5,70 ml). Ce volume de remplissage a été établi pendant le développement, et ce suremplissage permet après addition de tout le contenu du flacon de solvant au contenu du flacon de solution CABAZITAXEL REDDY PHARMA 60 mg, d’obtenir la concentration de la solution du prémélange de 10 mg/ml de CABAZITAXEL REDDY PHARMA.
6.6. Précautions particulières d’élimination et de manipulation
CABAZITAXEL REDDY PHARMA doit être préparé et manipulé seulement par un personnel formé à la manipulation des agents cytotoxiques. Les femmes enceintes ne doivent pas manipuler le produit. Comme tous les autres agents antinéoplasiques, des précautions doivent être prises pendant la manipulation et la préparation de la solution de CABAZITAXEL REDDY PHARMA, prenant en compte l’utilisation des dispositifs adaptés, des équipements de protection personnelle (comme des gants), et des procédures de préparation. En cas de contact cutané lors de chacune des étapes de préparation de CABAZITAXEL REDDY PHARMA, laver immédiatement et soigneusement la peau avec de l'eau et du savon. En cas de contact avec une muqueuse, laver immédiatement et soigneusement à grande eau la muqueuse contaminée.
Toujours diluer la solution à diluer avec la totalité du solvant fourni avant de l’ajouter dans la solution de perfusion.
Lisez attentivement la TOTALITE de la rubrique avant d’effectuer les étapes de mélange et de dilution. CABAZITAXEL REDDY PHARMA requiert DEUX dilutions avant administration. Suivez les instructions de préparation mentionnées ci-dessous.
Remarque : Le flacon de la solution à diluer de CABAZITAXEL REDDY PHARMA 60 mg/1,5 ml (volume de remplissage : 73,2 mg de cabazitaxel/1,83 ml) et le flacon de solvant (volume de remplissage : 5,70 ml) contiennent tous les deux un surremplissage pour compenser la perte de liquide survenant lors de la préparation.
Ce surremplissage permet d’obtenir une solution contenant 10 mg/ml de cabazitaxel après dilution de la solution à diluer de CABAZITAXEL REDDY PHARMA avec la TOTALITE du contenu du flacon de solvant fourni.
Les deux étapes suivantes concernant la procédure de dilution de la solution pour perfusion doivent être réalisées de manière aseptique.
|
Etape 1 : Dilution initiale de la solution à diluer avec le solvant fourni. |
|
|
Etape 1.1 |
|
|
Inspectez visuellement le flacon de la solution à diluer et le flacon de solvant fourni. La solution à diluer et le solvant doivent être limpides. |
|
|
|
|
|
Etape 1.2 |
|
|
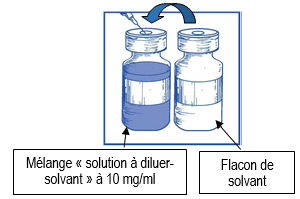
En utilisant une seringue munie d’une aiguille, prélevez de façon aseptique la totalité du contenu du flacon de solvant fourni en retournant partiellement le flacon. |
|
|
Etape 1.3 |
|
|
Injectez la totalité du contenu de la seringue dans le flacon de la solution à diluer correspondant. Afin de limiter autant que possible la formation de mousse en injectant le solvant, dirigez l’aiguille sur la paroi interne du flacon de la solution à diluer et injectez lentement. Une fois reconstituée, la solution obtenue contient 10 mg/ml de cabazitaxel. |
|
|
Etape 1.4 |
|
|
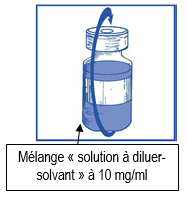
Retirez la seringue et l’aiguille et mélangez manuellement et doucement par retournements répétés de manière à obtenir une solution claire et homogène. Cela peut prendre environ 45 secondes. |
|
|
Etape 1.5 |
|
|
Laissez reposer cette solution environ 5 minutes puis vérifiez que la solution est homogène et claire. Il est normal que la mousse persiste après cette première étape. |
|
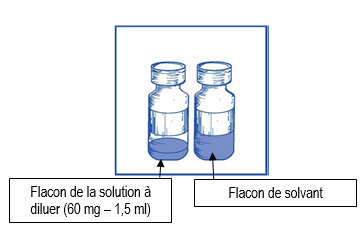
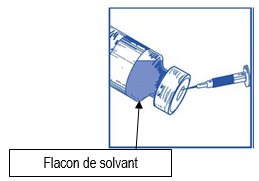
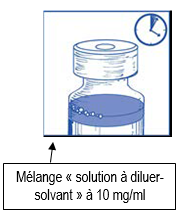
Ce mélange « solution à diluer-solvant » obtenu contient 10 mg/ml de cabazitaxel (au moins 6 ml de volume extractible). La seconde dilution doit être effectuée immédiatement (dans l’heure) comme détaillé dans l’étape 2.
Plus d’un flacon de mélange « solution à diluer-solvant » peut être nécessaire pour administrer la dose prescrite.
|
Etape 2 : Seconde dilution pour perfusion (dilution finale) |
|
|
Etape 2.1 |
|
|
Prélevez de façon aseptique le volume requis de mélange « solution à diluer-solvant » (contenant 10 mg/ml de cabazitaxel), avec une seringue graduée munie d’une aiguille. A titre d’exemple, une dose de 45 mg de CABAZITAXEL REDDY PHARMA nécessiterait 4,5 ml de mélange « solution à diluer-solvant » préparé selon les modalités de l’étape 1. Comme la mousse peut persister sur la paroi interne du flacon de cette solution à la suite de la préparation décrite à l’étape 1, il est préférable de placer l’aiguille de la seringue au milieu de la solution lors du prélèvement. |
|
|
Etape 2.2 |
|
|
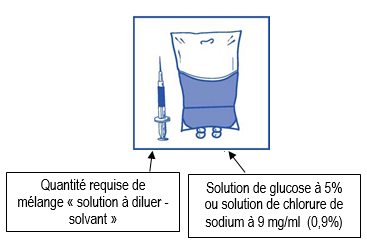
Injectez le contenu de la seringue dans une poche stérile pour perfusion exempte de PVC contenant soit une solution de glucose à 5% soit une solution de chlorure de sodium à 9 mg/ml (0,9%). La concentration de la solution à perfuser doit être comprise entre 0,10 mg/ml et 0,26 mg/ml. |
|
|
|
|
|
Etape 2.3 |
|
|
Retirez la seringue et mélanger le contenu de la poche ou flacon de perfusion par rotation manuelle. |
|
|
Etape 2.4 |
|
|
Comme tous les médicaments administrés par voie parentérale, la solution pour perfusion obtenue doit être contrôlée visuellement avant utilisation. Comme la solution pour perfusion est hypersaturée, elle peut parfois cristalliser avec le temps. Dans ce cas, la solution ne doit pas être utilisée et doit être détruite. |
|
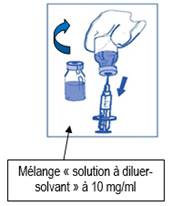

La solution pour perfusion doit être utilisée immédiatement. Toutefois, la durée de conservation peut être plus longue sous certaines conditions précisées dans la rubrique 6.3.
Un filtre en ligne de pores de 0,22 micromètre de diamètre (communément appelé 0,2 micromètre) est recommandé lors de l’administration.
N’utilisez pas des poches de perfusion en PVC ni de sets de perfusion contenant du polyuréthane pour la préparation et l’administration de CABAZITAXEL REDDY PHARMA.
CABAZITAXEL REDDY PHARMA ne doit pas être mélangé avec des médicaments autres que ceux mentionnés.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
9 AVENUE EDOUARD BELIN
92500 RUEIL-MALMAISON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 550 741 9 9 : 1,5 mL en flacon (verre) et 4,5 mL de solvant en flacon (verre).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
Date de première autorisation: 18 juin 2020
10. DATE DE MISE A JOUR DU TEXTE
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament réservé à l’usage hospitalier.
Prescription réservée aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 06/09/2022
CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion
Cabazitaxel
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant que CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion ne vous soit administré ?
3. Comment utiliser CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Agent antinéoplasiques. Code ATC : L01CD04.
CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion est utilisé pour traiter le cancer de la prostate qui a progressé après avoir eu une autre chimiothérapie. Il fonctionne en stoppant la croissance et la multiplication des cellules.
Vous prendrez aussi un autre médicament qui fait partie de votre traitement. Il s’agit d’un corticostéroïde (prednisone ou prednisolone) par voie orale tous les jours. Demandez à votre médecin qu’il vous donne des informations concernant cet autre médicament.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT QUE CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion NE VOUS SOIT ADMINISTRE ?
· vous êtes allergique (hypersensible) au cabazitaxel, aux autres taxanes, ou au polysorbate 80 ou à l’un des autres excipients contenus dans ce médicament, (mentionnés dans la rubrique 6).
· le nombre de vos globules blancs est trop bas (taux de neutrophiles inférieur ou égal à 1500 /mm3),
· vous avez une anomalie fonctionnelle modérée ou sévère du foie,
· vous avez récemment reçu ou vous êtes sur le point de recevoir un vaccin contre la fièvre jaune.
Vous ne devez pas prendre du CABAZITAXEL REDDY PHARMA si l’un de ces cas ci-dessus s’applique à vous. Si vous n’êtes pas sûr, parlez-en à votre médecin.
Avertissements et précautions
Avant chaque traitement par CABAZITAXEL REDDY PHARMA, vous aurez des examens sanguins pour vérifier que vous avez suffisamment de cellules sanguines et une fonction du foie et des reins suffisante pour recevoir CABAZITAXEL REDDY PHARMA.
Prévenez immédiatement votre médecin si:
· vous avez de la fièvre. Pendant le traitement avec CABAZITAXEL REDDY PHARMA, il est probable que votre taux de globules blancs diminue. Votre médecin suivra votre bilan sanguin et sera attentif à l’apparition de signes révélant une infection. Il/elle pourra ainsi vous prescrire des traitements pour maintenir votre taux de globules blancs. Les personnes ayant des taux de globules blancs bas peuvent développer des infections mettant en jeu le pronostic vital. Le premier signe précurseur d’infection étant la fièvre, si vous présentez ce signe, avertissez votre médecin immédiatement.
· vous avez déjà eu des allergies. Des réactions allergiques sévères peuvent apparaître pendant le traitement avec CABAZITAXEL REDDY PHARMA.
· vous avez des diarrhées sévères ou persistantes, vous avez envie de vomir (nausées) ou vous vomissez. Ces évènements peuvent provoquer une déshydratation sévère. Votre médecin pourra vous prescrire un traitement.
· vous avez une sensation d'engourdissement, de picotement, de brûlure ou de diminution de la sensation dans vos mains ou pieds.
· vous avez des problèmes de saignement de l’intestin ou vous avez un changement de la couleur de vos selles ou des douleurs d’estomac. Si le saignement ou la douleur est sévère, votre médecin arrêtera le traitement par CABAZITAXEL REDDY PHARMA. Car CABAZITAXEL REDDY PHARMA peut augmenter le risque de saignement ou de développer des trous dans la paroi intestinale.
· vous avez des problèmes rénaux.
· vous avez un jaunissement de la peau et des yeux, un assombrissement de l’urine, des nausées sévères (sensation d’être malade) ou vomissements, qui peuvent être les signes et symptômes de problèmes de foie.
· vous avez des problèmes de foie pendant le traitement.
· vous observez une augmentation ou diminution significative du volume urinaire quotidien.
· vous avez du sang dans les urines.
Si vous présentez l’une de ces situations cliniques, appelez votre médecin immédiatement. Votre médecin pourra réduire la dose de CABAZITAXEL REDDY PHARMA ou arrêter le traitement.
Enfants et adolescents
Sans objet.
Autres médicaments et CABAZITAXEL REDDY PHARMA
Si vous prenez ou avez pris récemment un autre médicament, y compris un médicament obtenu sans ordonnance, parlez-en à votre médecin, votre pharmacien ou votre infirmière. En effet d’autres médicaments peuvent affecter l’action de CABAZITAXEL REDDY PHARMA, de même que CABAZITAXEL REDDY PHARMA peut modifier l’action de ces médicaments. Ces médicaments incluent les suivants:
· kétoconazole, rifampicine (pour les infections) ;
· carbamazépine, phénobarbital ou phénytoïne (pour les crises d’épilepsie) ;
· millepertuis (Hypericum perforatum) (remède à base de plante pour la dépression et d’autres situations) ;
· statines (telles que simvastatine, lovastatine, atorvastatine, rosuvastatine ou pravastatine) (pour réduire le cholestérol dans le sang) ;
· valsartan (contre l’hypertension) ;
· répaglinide (contre le diabète).
Si vous devez vous faire vacciner durant votre traitement par CABAZITAXEL REDDY PHARMA, parlez-en à votre médecin.
CABAZITAXEL REDDY PHARMA avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
CABAZITAXEL REDDY PHARMA ne doit pas être utilisé chez les femmes enceintes ou en âge de procréer sans utilisation de contraception.
CABAZITAXEL REDDY PHARMA ne doit pas être utilisé durant l’allaitement.
Utilisez un préservatif pendant vos rapports sexuels si votre partenaire est ou peut devenir enceinte. CABAZITAXEL REDDY PHARMA peut être présent dans votre sperme et pourrait affecter le foetus. Il est déconseillé de concevoir un enfant pendant et jusqu’à 6 mois après le traitement et de prendre conseil pour la conservation du sperme avant le traitement car CABAZITAXEL REDDY PHARMA peut altérer la fertilité masculine.
Conduite de véhicules et utilisation de machines
Vous pourrez vous sentir fatigué ou avoir des vertiges en prenant ce médicament. Ne conduisez pas ou n’utilisez pas de machines avant de vous sentir mieux.
CABAZITAXEL REDDY PHARMA contient de l’alcool (éthanol)
Ce médicament contient 573 mg d’alcool (éthanol) dans chaque flacon de solvant. La quantité contenue dans la dose de ce médicament est équivalente à moins de 15 ml de bière ou 6 ml de vin. La faible quantité d’alcool dans ce médicament n’aura pas d’effet notable.
3. COMMENT UTILISER CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion ?
Des médicaments antiallergiques vous seront donnés avant votre traitement par CABAZITAXEL REDDY PHARMA afin de réduire les risques de réactions allergiques.
· CABAZITAXEL REDDY PHARMA vous sera administré par un médecin ou une infirmière.
· CABAZITAXEL REDDY PHARMA doit être préparé (dilué) avant d’être administré. Les informations pratiques pour la préparation et l’administration de CABAZITAXEL REDDY PHARMA destinées aux médecins, infirmières et pharmaciens sont fournies dans cette notice.
· CABAZITAXEL REDDY PHARMA vous sera administré par injection (perfusion) dans une de vos veines (voie intraveineuse). La perfusion durera environ 1 heure pendant laquelle vous serez à l’hôpital.
· Dans le cadre de votre traitement, vous prendrez également un médicament corticoïde (prednisone ou prednisolone) par voie orale chaque jour.
Combien et à quelle fréquence
· La dose habituelle dépend de votre surface corporelle. Votre médecin calculera votre surface corporelle en mètres carrés (m2) et déterminera la dose qu’il vous convient de vous administrer.
· Habituellement vous recevrez une perfusion toutes les trois semaines.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien hospitalier ou à votre infirmière.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Votre médecin vous en parlera et vous expliquera les risques et bénéfices potentiels de votre traitement.
Consultez un médecin immédiatement si vous notez un de ces effets indésirables :
· fièvre (température élevée). Cet effet est t fréquent (pouvant survenir chez 1 personne sur 10 au maximum).
· perte importante de liquide corporel (déshydratation). Cet effet est fréquent (pouvant survenir chez 1 personne sur 10 au maximum) et peut arriver si vous avez une diarrhée importante ou persistante, ou de la fièvre, ou si vous êtes malade (vomissement).
· des douleurs sévères de l’estomac, ou des douleurs de l’estomac persistantes. Ceci peut arriver si vous avez un trou dans l’estomac, l’oesophage ou l’intestin (perforation digestive). Ceci peut entraîner la mort.
Si vous ressentez un de ces effets, prévenez immédiatement votre médecin.
Autres effets indésirables:
Très fréquent (pouvant survenir chez plus d’1 personne sur 10) :
· diminution du nombre de globules rouges (anémie) ou de globules blancs (qui jouent un rôle important contre les infections)
· diminution du nombre de plaquettes, (qui entraine un risque élevé de saignement)
· perte de l’appétit (anorexie)
· troubles de la digestion, troubles gastriques incluant nausées, vomissements, diarrhée ou constipation
· douleur dorsale
· sang dans les urines
· sensation de fatigue, faiblesse ou manque d’énergie
Fréquent (pouvant survenir chez 1 personne sur 10 au maximum):
· altération du goût
· respiration courte
· toux
· douleur abdominale
· perte temporaire des cheveux (dans la plupart des cas, les cheveux repousseront normalement à l’arrêt du traitement)
· douleur des articulations
· infection urinaire
· manque de globules blancs associé à de la fièvre et une infection
· sensation d’engourdissement, de picotement, de brûlure ou la perte de sensibilité des mains et des pieds
· étourdissements
· maux de tête
· diminution ou augmentation de la tension artérielle
· impression de lourdeur dans l’estomac, brûlure d’estomac ou éructation
· douleur gastrique
· hémorroïdes
· spasme musculaire
· miction douloureuse ou fréquente
· incontinence urinaire
· maladie rénale ou problèmes aux reins
· plaie buccale ou sur les lèvres
· infections ou risque d’infections
· taux de sucre élevé dans le sang
· insomnie
· confusion mentale
· sensation d’anxiété
· sensation anormale ou perte de sensation ou douleur dans les bras et les jambes
· trouble de l’équilibre
· rythme cardiaque rapide ou irrégulier
· caillot de sang dans la jambe ou le poumon
· sensation de rougissement de la peau
· douleur dans la bouche ou la gorge
· saignement rectal
· inconfort, douleurs, ou faiblesse musculaires
· gonflement des pieds ou des jambes
· frissons
· altération des ongles (changement de la couleur des ongles ; ongles qui pourraient se détacher).
Peu fréquent (peut affecter jusqu’à 1 personne sur 100) :
· taux de potassium bas dans le sang
· bourdonnement dans les oreilles
· sensation de chaleur dans la peau
· rougeur de la peau
· inflammation de la vessie, qui peut se produire lorsque votre vessie a été précédemment exposée à la radiothérapie (cystite due à un phénomène de rappel après radiothérapie).
Fréquence inconnue (ne peut être estimée à partir des données disponibles) :
· pneumopathie interstitielle diffuse (inflammation des poumons entraînant une toux et des
· difficultés respiratoires).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant pour solution pour perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étui et l’étiquette des flacons après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Ne pas mettre au réfrigérateur.
Les informations sur la conservation et la durée d’utilisation de CABAZITAXEL REDDY PHARMA, une fois qu’il a été dilué et qu’il est prêt à l’utilisation, sont décrites dans la rubrique « GUIDE DE PRÉPARATION, ADMINISTRATION ET MANIPULATION DE CABAZITAXEL REDDY PHARMA A USAGE DES PROFESSIONNELS DE SANTE ».
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Cabazitaxel...................................................................................................................... 40 mg
Pour 1ml de solution à diluer.
Chaque flacon à diluer contient 60 mg de cabazitaxel.
· Les autres composants sont : polysorbate 80, acide citrique (pour la solution à diluer), éthanol à 96% et eau pour préparations injectables (pour le solvant), voir rubrique CABAZITAXEL REDDY PHARMA contient de l’alcool.
Remarque : le flacon de solution à diluer de CABAZITAXEL REDDY PHARMA 60 mg/1,5 ml (volume de remplissage : 73,2 mg de cabazitaxel/1,83 ml) et le flacon de solvant (volume de remplissage : 5,70 ml) contiennent tous les deux un surremplissage pour compenser la perte de liquide survenant lors de la préparation.
Ce surremplissage permet d’obtenir une solution contenant 10 mg/ml de cabazitaxel après dilution de la solution à diluer de CABAZITAXEL REDDY PHARMA avec la TOTALITE du contenu du flacon de solvant fourni.
Qu’est-ce que CABAZITAXEL REDDY PHARMA et contenu de l’emballage extérieur
CABAZITAXEL REDDY PHARMA est une solution à diluer et solvant pour perfusion (solution à diluer stérile).
La solution à diluer est une solution huileuse, limpide, jaune à jaune marron.
Le solvant est une solution limpide et légèrement jaune.
Chaque boîte de CABAZITAXEL REDDY PHARMA contient :
· Un flacon rond en verre à usage unique de 15 ml, fermé par un bouchon en caoutchouc scellé par une capsule de type flip-off, contenant 1,5 ml (volume nominal) de solution à diluer.
· Un flacon rond en verre à usage unique de 15 ml, fermé par un bouchon en caoutchouc scellé par une capsule de type flip-off, contenant 4,5 ml (volume nominal) de solvant.
Titulaire de l’autorisation de mise sur le marché
9 AVENUE EDOUARD BELIN
92500 RUEIL-MALMAISON
Exploitant de l’autorisation de mise sur le marché
REDDY PHARMA SAS
9 AVENUE EDOUARD BELIN
92500 RUEIL-MALMAISON
KOBELWEG 95
86156 AUGSBURG
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
Allemagne : Cabazitaxel beta 60 mg Konzentrat und Lösungsmittel zur Herstellung einer Infusionslösung
France : Cabazitaxel Reddy Pharma 60 mg solution à diluer et solvant pour solution pour perfusion
Espagne : Cabazitaxel Dr. Reddys concentrado y dispolvente para solución para perfusión EFG
Italie : Cabazitaxel Dr. Reddy´s concentrato e solvente per soluzione per infusione
Royaume-uni : Cabazitaxel beta 60 mg concentrate and solvent for solution for infusion
La dernière date à laquelle cette notice a été révisée est :
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
----------------------------------------------------------------------------------------------------------------------------------------
Les informations suivantes sont destinées exclusivement aux professionnels de santé:
GUIDE DE PRÉPARATION, ADMINISTRATION ET MANIPULATION DE CABAZITAXEL REDDY PHARMA 60 mg SOLUTION A DILUER ET SOLVANT POUR SOLUTION POUR PERFUSION A USAGE DES PROFESSIONNELS DE SANTE:
Ces informations complètent les rubriques 3 et 5 pour l’utilisateur.
Il est important que vous lisiez la totalité de cette procédure avant la préparation de la solution pour perfusion.
Incompatibilités
Ce médicament ne doit pas être mélangé avec d’autres médicaments à l’exception de ceux utilisés pour les dilutions. Les poches de perfusion en PVC ou les sets de perfusion en polyuréthane ne doivent pas être utilisées pour la préparation et l’administration de la solution de perfusion.
Durée de conservation et précautions spéciales de conservation
Pour les boîtes de CABAZITAXEL REDDY PHARMA 60 mg solution à diluer et solvant
Ne pas mettre au réfrigérateur.
Après ouverture
Les flacons de solution et de solvant doivent être utilisés immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l’utilisateur. D’un point de vue microbiologique, la procédure de dilution en 2 étapes doit être réalisée dans des conditions d’asepsie contrôlées et validées (voir ci-dessous « Précautions pour la préparation et l’administration »).
Après la dilution initiale de CABAZITAXEL REDDY PHARMA 60 mg solution à diluer avec la totalité du contenu du flacon de solvant, la stabilité physico-chimique a été démontrée pendant 1 heure à température ambiante et pendant 21 jours à 2°C-8°C.
D’un point de vue microbiologique, la solution pour perfusion doit être utilisée immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l’utilisateur et ne devraient pas normalement dépasser 24 heures à 2°C- 8°C, sauf en cas de dilution réalisée dans des conditions d’asepsie contrôlées et validées.
Après la dilution finale dans le flacon/poche pour perfusion
La stabilité physico-chimique de la solution pour perfusion a été démontrée pendant 8 heures à température ambiante (15°C - 30°C) incluant l’heure d’administration de la perfusion et pendant 48 heures au réfrigérateur (2°C-8°C) incluant l’heure d’administration de la perfusion dans la poche de perfusion et de 8 heures à température ambiante (15°C-30°C) incluant l’heure d’administration dans le flacon en verre.
D’un point de vue microbiologique, la solution pour perfusion doit être utilisée immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l’utilisateur et ne devraient pas normalement dépasser 24 heures à 2°C- 8°C, sauf en cas de dilution réalisée dans des conditions d’asepsie contrôlées et validées.
Précautions pour la préparation et l’administration
Comme tous les autres agents antinéoplasiques, des précautions doivent être prises pendant la manipulation et la préparation de solutions de CABAZITAXEL REDDY PHARMA, prenant en compte des dispositifs adaptés, des équipements de protection personnelle (comme des gants), et des procédures de préparation.
En cas de contact cutané lors de chacune des étapes de préparation de CABAZITAXEL REDDY PHARMA, lavez immédiatement et soigneusement la peau avec de l'eau et du savon. En cas de contact avec une muqueuse, lavez immédiatement et soigneusement à grande eau la muqueuse contaminée.
CABAZITAXEL REDDY PHARMA doit être préparé et administré uniquement par un personnel formé et habilité à la manipulation d’agents cytotoxiques. Aucune personne enceinte ne devra manipuler ce médicament.
Toujours diluer la solution à diluer avec la totalité du solvant fourni avant de l’ajouter dans la solution de perfusion.
Étapes de préparation
Lisez attentivement la TOTALITE de la rubrique avant d’effectuer les étapes de mélange et de dilution. CABAZITAXEL REDDY PHARMA requiert DEUX dilutions avant administration. Suivez les instructions de préparation mentionnées ci-dessous.
Remarque : Le flacon de solution à diluer de CABAZITAXEL REDDY PHARMA 60 mg/1,5 ml (volume de remplissage : 73,2 mg de cabazitaxel/1,83 ml) et le flacon de solvant (volume de remplissage : 5,70 ml) contiennent tous les deux un surremplissage pour compenser la perte de liquide survenant lors de la préparation.
Ce surremplissage permet d’obtenir une solution contenant 10 mg/ml de cabazitaxel après dilution de la solution à diluer de CABAZITAXEL REDDY PHARMA avec la TOTALITE du contenu du flacon de solvant fourni.
Les deux étapes suivantes concernant la procédure de dilution de la solution pour perfusion doivent être réalisées de manière aseptique.
|
Etape 1 : Dilution initiale de la solution à diluer avec le solvant fourni. |
|
|
Etape 1.1 |
|
|
Inspectez visuellement le flacon de la solution à diluer et le flacon de solvant fourni. La solution à diluer et le solvant doivent être limpides. |
|
|
|
|
|
Etape 1.2 |
|
|
En utilisant une seringue munie d’une aiguille, prélevez de façon aseptique la totalité du contenu du flacon de solvant fourni en retournant partiellement le flacon. |
|
|
Etape 1.3 |
|
|
Injectez la totalité du contenu de la seringue dans le flacon de la solution à diluer correspondant. Afin de limiter autant que possible la formation de mousse en injectant le solvant, dirigez l’aiguille sur la paroi interne du flacon de la solution à diluer et injectez lentement. Une fois reconstituée, la solution obtenue contient 10 mg/ml de cabazitaxel. |
|
|
Etape 1.4 |
|
|
Retirez la seringue et l’aiguille et mélangez manuellement et doucement par retournements répétés de manière à obtenir une solution claire et homogène. Cela peut prendre environ 45 secondes. |
|
|
Etape 1.5 |
|
|
Laissez reposer cette solution environ 5 minutes puis vérifiez que la solution est homogène et claire. Il est normal que la mousse persiste après cette première étape. |
|
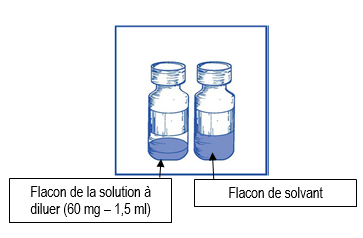
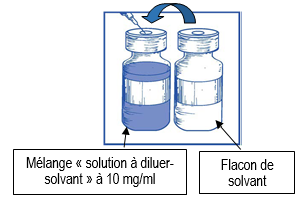
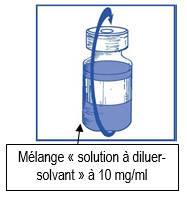
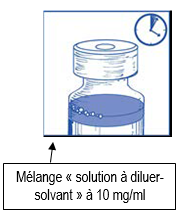
Ce mélange « solution à diluer-solvant » obtenu contient 10 mg/ml de cabazitaxel (au moins 6 ml de volume extractible). La seconde dilution doit être effectuée immédiatement (dans l’heure) comme détaillé dans l’étape 2.
Plus d’un flacon de mélange « solution à diluer-solvant » peut être nécessaire pour administrer la dose prescrite.
|
Etape 2 : Seconde dilution pour perfusion (dilution finale) |
|
|
Etape 2.1 |
|
|
Prélevez de façon aseptique le volume requis de mélange « solution à diluer-solvant » (contenant 10 mg/ml de cabazitaxel), avec une seringue graduée munie d’une aiguille. A titre d’exemple, une dose de 45 mg de CABAZITAXEL REDDY PHARMA nécessiterait 4,5 ml de mélange « solution à diluer-solvant » préparé selon les modalités de l’étape 1. Comme la mousse peut persister sur la paroi interne du flacon de cette solution à la suite de la préparation décrite à l’étape 1, il est préférable de placer l’aiguille de la seringue au milieu de la solution lors du prélèvement. |
|
|
Etape 2.2 |
|
|
Injectez le contenu de la seringue dans une poche stérile pour perfusion exempte de PVC contenant soit une solution de glucose à 5% soit une solution de chlorure de sodium à 9 mg/ml (0,9%). La concentration de la solution à perfuser doit être comprise entre 0,10 mg/ml et 0,26 mg/ml. |
|
|
|
|
|
Etape 2.3 |
|
|
Retirez la seringue et mélanger le contenu de la poche ou flacon de perfusion par rotation manuelle. |
|
|
Etape 2.4 |
|
|
Comme tous les médicaments administrés par voie parentérale, la solution pour perfusion obtenue doit être contrôlée visuellement avant utilisation. Comme la solution pour perfusion est hypersaturée, elle peut parfois cristalliser avec le temps. Dans ce cas, la solution ne doit pas être utilisée et doit être détruite. |
|

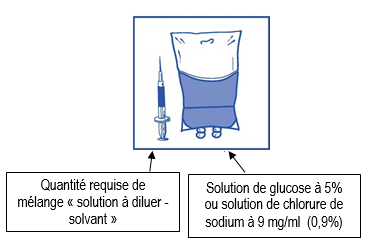

La solution pour perfusion doit être utilisée immédiatement. Toutefois, la durée de conservation peut être plus longue sous certaines conditions précisées dans la rubrique ci-dessus Durée de conservation et précautions spéciales de conservation.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Méthode d’administration
CABAZITAXEL REDDY PHARMA est administré en perfusion pendant une heure.
Un filtre en ligne de pores de 0,22 micromètre de diamètre (également appelé 0,2 micromètre) est recommandé lors de l’administration.
Les poches de perfusion en PVC ou les sets de perfusion en polyuréthane ne doivent pas être utilisées pour la préparation et l’administration de la solution de perfusion.