Dernière mise à jour le 29/06/2026
TEGLUTIK 5 mg/ml, suspension buvable
Indications thérapeutiques
Classe pharmacothérapeutique : autres médicaments du système nerveux, code ATC : N07XX02.
TEGLUTIK est un médicament qui agit sur le système nerveux.
Dans quel cas TEGLUTIK est-il utilisé :
TEGLUTIK est prescrit chez des patients avec une sclérose latérale amyotrophique (SLA).
La SLA est une forme de maladie du motoneurone qui affecte les cellules nerveuses responsables de l'envoi d'instructions aux muscles, ce qui entraîne une faiblesse musculaire, une atrophie musculaire et une paralysie.
La destruction des cellules nerveuses dans les maladies du motoneurone peut être provoquée par une quantité trop importante de glutamate (un messager chimique) dans le cerveau et la moelle épinière. TEGLUTIK bloque la libération de glutamate, ce qui peut contribuer à empêcher la destruction des cellules nerveuses.
Veuillez consulter votre médecin pour des informations complémentaires sur la SLA et les raisons pour lesquelles ce médicament vous a été prescrit.
Présentations
> 1 flacon(s) en verre jaune(brun) de 300 ml avec fermeture de sécurité avec adaptateur polyéthylène basse densité (PEBD) avec seringue(s) pour administration orale polyéthylène haute densité (PEHD)
Code CIP : 275 660-1 ou 34009 275 660 1 1
Déclaration de commercialisation : 05/04/2016
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 75,83 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 76,85 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 16/09/2015 | Inscription (CT) | le service médical rendu par TEGLUTIK est important dans l’indication de l’AMM, au même titre que RILUTEK. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 16/09/2015 | Inscription (CT) | TEGLUTIK (riluzole sous forme de suspension buvable) est un médicament hybride qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à la spécialité de référence RILUTEK (riluzole sous forme comprimé) et ses génériques. |
Autres informations
- Titulaire de l'autorisation : ITALFARMACO SA
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription initiale annuelle réservée à certains spécialistes
- prescription réservée aux spécialistes et services NEUROLOGIE
- renouvellement non restreint
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 841 359 6
ANSM - Mis à jour le : 19/02/2026
TEGLUTIK 5 mg/ml, suspension buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
riluzole.................................................................................................................................... 5 mg
Pour 1 ml de suspension buvable
Excipient(s) à effet notoire : 1 ml de suspension buvable contient 400 mg de sorbitol E420 (équivalent à 571,43 mg de sorbitol liquide (70% m/m).
Pour la liste complète des excipients, voir rubrique 6.1.
Suspension homogène légèrement brune et opaque après agitation manuelle.
4.1. Indications thérapeutiques
Les essais cliniques ont montré que le riluzole augmente la survie des patients atteints de SLA (voir rubrique 5.1).
La définition de la survie était : patient vivant, non intubé pour ventilation mécanique assistée et non trachéotomisé.
Aucune action thérapeutique de TEGLUTIK sur les fonctions motrices, la fonction respiratoire, les fasciculations, la force musculaire et les symptômes moteurs n’a été mise en évidence. TEGLUTIK n’a pas montré d’effet bénéfique aux stades avancés de la SLA.
L’efficacité et la sécurité d’emploi de TEGLUTIK n’ont été étudiées que dans la SLA. Par conséquent, le riluzole ne doit pas être utilisé chez les patients atteints d’une autre forme de maladie du motoneurone.
4.2. Posologie et mode d'administration
Posologie
La posologie quotidienne recommandée chez l'adulte ou la personne âgée est de 100 mg (50 mg toutes les douze heures). Aucun bénéfice supplémentaire significatif ne peut être attendu à posologie supérieure.
Il est recommandé de prendre 10 ml de suspension deux fois par jour (10 ml de suspension correspondent à 50 mg de riluzole).
Populations à risque
Population pédiatrique
L’usage de TEGLUTIK n’est pas recommandé dans la population pédiatrique, en l’absence de données sur l'efficacité et la sécurité d’emploi du riluzole dans les maladies neurodégénératives de l'enfant ou de l'adolescent.
Insuffisants rénaux
L’usage de TEGLUTIK n’est pas recommandé chez les insuffisants rénaux en raison de l’absence d’étude à dose répétée réalisée chez ce type de patients (voir rubrique 4.4).
Personnes âgées :
Compte tenu des données pharmacocinétiques, il n’y a pas de recommandation particulière pour l’utilisation de TEGLUTIK dans cette population.
Patients avec insuffisance hépatique :
(voir rubrique 4.3, rubrique 4.4, et rubrique 5.2).
Mode d’administration
La suspension peut être administrée par voie orale ou également par sonde de gastrostomie. Une dilution dans un liquide n'est pas nécessaire.
La suspension s'administre au moyen d'une seringue graduée.
Se reporter à la rubrique 6.6 pour des instructions sur la manipulation du produit.
· Hypersensibilité à la substance active ou à l'un des excipients mentionnés dans la rubrique 6.1.
· Maladie hépatique ou taux de transaminases supérieur à 3 fois la limite supérieure de la normale avant la mise en route du traitement.
· Femme enceinte ou allaitante
4.4. Mises en garde spéciales et précautions d'emploi
Le riluzole doit être utilisé avec prudence chez les patients ayant des antécédents de dysfonctionnement hépatique, ou chez les patients présentant une légère élévation des transaminases sériques (ALAT/SGPT; ASAT/SGOT jusqu’à 3 fois la limite supérieure de la normale), de la bilirubine et/ou des gamma-glutamyl transférases (GGT). Une perturbation de plusieurs paramètres hépatiques (en particulier taux de bilirubine élevé) doit faire déconseiller l’utilisation de riluzole (voir rubrique 4.8).
Du fait du risque d’hépatite, le taux de transaminases sériques, dont les ALAT (SGPT), doit être contrôlé avant la mise sous traitement et pendant la durée du traitement par le riluzole. Les ALAT doivent être dosées tous les mois pendant les 3 premiers mois, puis tous les 3 mois pendant la première année et périodiquement ensuite. Ce suivi devra être plus fréquent chez les patients dont le taux d’ALAT s'élève sous traitement.
Le traitement par le riluzole devra être interrompu si les taux d’ALAT augmentent jusqu’à 5 fois la limite supérieure de la normale ou au-delà. Les effets d'une réduction posologique ou d'une réadministration ultérieure chez les patients dont le taux de ALAT a atteint ou dépassé 5 fois la limite supérieure de la normale, ne sont pas connus.
Toute réadministration du riluzole chez ces patients est donc déconseillée.
Neutropénie
Les patients doivent être avertis qu’il convient d’informer leur médecin de toute maladie fébrile. La survenue d’une maladie fébrile doit entraîner un contrôle de la numération formule sanguine et une interruption du traitement par riluzole en cas de neutropénie (voir rubrique 4.8).
Pneumopathie interstitielle
Des cas de pneumopathie interstitielle, dont certains sévères, ont été rapportés chez des patients traités par le riluzole (voir rubrique 4.8). Une radiographie pulmonaire doit être effectuée en cas d'apparition de symptômes respiratoires tels qu’une toux sèche et/ou une dyspnée, et l'administration du riluzole doit être arrêtée immédiatement si des aspects suggèrent une pneumopathie interstitielle (opacités pulmonaires diffuses bilatérales par exemple). Dans la majorité des cas rapportés, les symptômes ont disparu après l’arrêt du médicament et un traitement symptomatique.
Insuffisance rénale
Aucune étude à dose répétée n’a été réalisée chez les patients ayant une insuffisance rénale (voir rubrique 4.2).
Excipients :
Ce médicament contient 4000 mg de sorbitol (E420) par dose de 10 ml de suspension buvable. L’effet additif des produits administrés concomitamment contenant du sorbitol (ou du fructose) et l’apport alimentaire de sorbitol (ou de fructose) doit être pris en compte.
La teneur en sorbitol dans les médicaments à usage oral peut affecter la biodisponibilité d’autres médicaments à usage oral administrés de façon concomitante.
Les patients présentant une intolérance héréditaire au fructose (IHF) ne doivent pas prendre ce médicament..
Ce médicament contient moins de 1 mmol (23 mg) par dose de 10 ml, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Il n'y a pas eu d'études cliniques évaluant les interactions du riluzole avec d'autres médicaments.
Des études in vitro utilisant des préparations à base de microsomes hépatiques humains suggèrent que le CYP 1A2 est le principal isoenzyme impliqué dans le métabolisme oxydatif initial du riluzole. Les inhibiteurs du CYP 1A2 (dont la caféine, le diclofenac, le diazépam, la nicergoline, la clomipramine, l’imipramine, la fluvoxamine, la phénacétine, la théophylline, l’amitriptyline et les quinolones) peuvent potentiellement diminuer le taux d’élimination du riluzole, tandis que les inducteurs du CYP 1A2 (dont la fumée de cigarette, la nourriture fumée au charbon de bois, la rifampicine et l’oméprazole) pourraient augmenter le taux d’élimination du riluzole.
4.6. Fertilité, grossesse et allaitement
Grossesse
TEGLUTIK est contre-indiqué en cas de grossesse (voir rubriques 4.3 et 5.3). Il n’y a pas d’expérience clinique d’utilisation du riluzole chez la femme enceinte.
TEGLUTIK est contre-indiquée chez la femme qui allaite (voir rubriques 4.3 et 5.3). Le passage du riluzole dans le lait maternel humain n'est pas connu.
Fertilité
Les études de fertilité chez le rat ont mis en évidence une légère altération des fonctions de reproduction et de la fertilité aux doses de 15 mg/kg/jour (supérieure à la dose thérapeutique), ceci étant probablement dû aux effets de sédation et de léthargie.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets sur l'aptitude à conduire des véhicules et à utiliser des machines n’ont pas été étudiés.
Résumé du profil de sécurité
Dans les essais cliniques de phase III conduits chez les patients atteints de SLA et traités par riluzole, les effets indésirables les plus fréquemment rapportés ont été l’asthénie, les nausées et les anomalies des paramètres biologiques hépatiques.
Résumé tabulé des effets indésirables
Les effets indésirables sont indiqués ci-dessous et classés selon la fréquence définie par la convention suivante : très fréquent (≥1/10), fréquent (≥1/100 à <1/10), peu fréquent (≥1/1000 à <1/100), rare (≥1/10 000 à <1/1000), très rare (<1/10 000), fréquence indéterminée (ne peut être pas estimée sur la base des données disponibles).
|
|
Très fréquent |
Fréquent |
Peu fréquent |
Fréquence indéterminée |
|
Affections hématologiques et du système lymphatique |
Anémie |
Neutropénie sévère (voir rubrique 4.4) |
||
|
Affections du système immunitaire |
Réaction de type anaphylactique, angioedème |
|||
|
Affections du système nerveux |
céphalées, étourdissements, paresthésie buccale et somnolence |
|||
|
Affections cardiaques |
Tachycardie |
|||
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Pneumopathie interstitielle (voir rubrique 4.4) |
|
|
Affections gastro-intestinales |
nausées |
diarrhée, douleur abdominale, vomissements |
pancréatite |
|
|
Affection de la peau et du tissu sous-cutané |
|
|
|
Éruption cutanée |
|
Affections hépatobiliaires |
anomalies des paramètres biologiques hépatiques |
|
|
Hépatite |
|
Troubles généraux et anomalies au site d’administration |
asthénie |
douleur |
|
|
Description des effets indésirables
Affections hépato-biliaires
L’élévation des alanineaminotransférases apparaissait généralement dans les trois premiers mois du traitement par riluzole. Elle a été habituellement transitoire et leur niveau est revenu à une valeur inférieure à 2 fois la limite supérieure de la normale après 2 à 6 mois malgré la poursuite du traitement. Ces élévations peuvent être associées à un ictère. Chez les patients des études cliniques (n=20) présentant une élévation des ALAT supérieure à 5 fois la limite supérieure de la normale (LSN), le traitement a été interrompu et leur niveau est revenu à une valeur inférieure à 2 fois la LSN dans les 2 à 4 mois dans la plupart des cas (voir rubrique 4.4).
Les données d’études indiquent que les patients asiatiques peuvent être plus susceptibles de présenter des anomalies des tests biologiques hépatiques- 3,2% (194/5995) des patients asiatiques et 1,8% (100/5641) des patients caucasiens.
Riluzole en suspension buvable
L'exposition totale au riluzole en suspension buvable et en comprimés était bioéquivalente, mais la Cmax moyenne (la concentration plasmatique maximale en riluzole après ingestion) du riluzole en suspension buvable était environ 20 % plus élevée que la Cmax moyenne du riluzole en comprimés. (voir rubrique 5.2).
Il pourrait y avoir un risque légèrement plus élevé d'événements indésirables, (par ex. vertiges, diarrhée, asthénie et augmentation de l'ALAT) avec la suspension buvable.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
Des cas isolés de symptômes psychiatriques et neurologiques, encéphalopathie toxique aiguë s’accompagnant d’un état stuporeux, coma et méthémoglobinémie ont été observés.
En cas de surdosage, le traitement est symptomatique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : autres médicaments du système nerveux, code ATC : N07XX02.
Mécanisme d’action
Bien que la pathogénèse de la SLA ne soit pas totalement élucidée, il semble que le glutamate (principal neurotransmetteur excitateur du système nerveux central) joue un rôle dans la mort cellulaire liée à la maladie.
Le riluzole agirait par inhibition de processus glutamatergiques. Le mécanisme d’action est incertain.
Efficacité et sécurité clinique
Dans un essai randomisé, 155 patients ont reçu soit du riluzole à la dose de 100 mg/jour (50 mg deux fois par jour), soit du placebo et ont été suivis pendant une durée de 12 à 21 mois. La survie, telle que définie ci-dessus (second paragraphe de la rubrique 4.1), a été significativement augmentée chez les patients recevant le riluzole comparativement au groupe placebo. La médiane de survie a été de 17,7 mois pour les patients recevant le riluzole contre 14,9 mois pour le groupe placebo.
Dans une étude dose-réponse, 959 patients atteints de SLA ont été randomisés en quatre groupes : riluzole 50, 100 ou 200 mg/jour et placebo, et ont été suivis pendant 18 mois. Chez les patients traités par 100 mg/jour de riluzole, la survie a été significativement augmentée par rapport à celle des patients recevant le placebo. L'effet du riluzole à la dose de 50 mg/jour n'a pas été statistiquement différent du placebo, l’effet du riluzole à la dose de 200 mg/jour a été comparable à l’effet observé avec 100 mg/jour. La médiane de survie a été d’environ 16,5 mois pour les patients recevant le riluzole 100 mg/jour contre 13,5 mois pour le groupe placebo.
Dans un essai en groupe parallèle, réalisé en vue d’évaluer l’efficacité et la tolérance du riluzole chez des malades à un stade avancé de la maladie, la durée de survie et les fonctions motrices n’ont pas été significativement différentes dans le groupe recevant le riluzole et dans le groupe placebo. Dans cette étude, la majorité des patients avaient une capacité vitale inférieure à 60 %.
Dans un essai randomisé en double-aveugle contre placebo, réalisé en vue d'évaluer l'efficacité et la tolérance du riluzole chez des patients japonais, 204 malades ont reçu soit du riluzole à la dose de 100 mg/ jour (50 mg deux fois par jour), soit du placebo et ont été suivis pendant 18 mois. Dans cette étude, l'efficacité a été évaluée sur l'incapacité à se déplacer seul, sur l'atteinte fonctionnelle des membres supérieurs, la trachéotomie, le recours à la ventilation assistée, l'alimentation par sonde gastrique ou la mort. La survie sans trachéotomie dans le groupe de patients traités par riluzole n'a pas été significativement différente de celle du groupe placebo.
Cependant, la puissance de cette étude à détecter des différences entre les groupes de traitements était faible. Une méta-analyse incluant cette étude et celles décrites plus haut montre un effet du riluzole par rapport au placebo moins marqué sur la survie, mais les différences sont toujours statistiquement significatives.
5.2. Propriétés pharmacocinétiques
Les propriétés pharmacocinétiques du riluzole ont été évaluées après administration orale d'une dose unique allant de 25 à 300 mg et après administration orale réitérée de doses allant de 25 mg à 100 mg deux fois par jour chez des volontaires sains de sexe masculin.
Les concentrations plasmatiques augmentent de façon linéaire avec la dose et le profil cinétique est indépendant de la dose. Après administration répétée (50 mg de riluzole 2 fois par jour pendant 10 jours), la concentration plasmatique du riluzole inchangé double et l'état d’équilibre est atteint en moins de 5 jours.
Le riluzole est rapidement absorbé après administration orale et les concentrations plasmatiques maximales sont atteintes en 60 à 90 minutes (Cmax = 173 ± 72 (DS) ng/ml). Environ 90 % de la dose sont absorbés. La biodisponibilité absolue du riluzole est de 60 % ± 18 %.
La prise de nourriture riche en graisses réduit la vitesse et le niveau d'absorption du riluzole (diminution de la Cmax de 44 % et de l'aire sous la courbe de 17 %).
Une étude de bioéquivalence a montré que l'exposition au riluzole en comprimés à 50 mg était équivalente à celle du riluzole en suspension buvable à 5 mg/ml. (Rapport 106,84 % ; IC 90 % 96,98– 117,71 %). Le riluzole est plus rapidement absorbé après administration de suspension buvable (Tmax d'environ 30 minutes), avec une Cmax environ 20 % supérieure à celle de l'administration de riluzole en comprimés (Rapport 122,32 % ; IC 90 % 103,28– 144,88 %) (voir rubrique 4.8).
Distribution
Le riluzole se distribue largement dans l'ensemble de l’organisme et traverse la barrière hématoencéphalique.
Son volume de distribution est d’environ 245 ± 69 l (3,4 l/kg). La liaison aux protéines plasmatiques est d'environ 97 %, le riluzole étant lié essentiellement à l'albumine sérique et aux lipoprotéines.
Biotransformation
Le riluzole sous forme inchangée est le composant principal retrouvé dans le plasma. Il est fortement métabolisé par le cytochrome P 450 puis subit une glucuronidation. Des études in vitro sur préparations de cellules de foie humain ont montré que le cytochrome P 450 1A2 est la principale isoenzyme impliquée dans le métabolisme du riluzole. Les métabolites identifiés dans les urines sont trois dérivés phénoliques et un dérivé uréido. Du riluzole inchangé est également retrouvé.
La voie metabolique principale du riluzole est une oxydation initiale par le cytochrome P 450 1A2, donnant le N-hydroxy-riluzole (RPR112512), principal métabolite actif du riluzole. Ce métabolite est rapidement glucuronoconjugué en dérivés O- et N- glucuronides.
Élimination
La demi-vie d'élimination est de 9 à 15 heures. Le riluzole est éliminé principalement dans les urines.
L'excrétion urinaire totale représente environ 90 % de la dose. Les glucuronides représentent plus de 85 % des métabolites retrouvés dans les urines. Seulement 2 % d’une dose de riluzole sont retrouvés sous forme inchangée dans les urines.
Populations à risque
Insuffisants rénaux : après administration orale d’une dose unique de 50 mg de riluzole, il n’y a pas de différence significative entre les paramètres cinétiques obtenus chez des patients insuffisants rénaux modérés ou sévères (clairance de la créatinine comprise entre 10 et 50 ml.min-1) et des volontaires sains.
Personnes âgées : les paramètres pharmacocinétiques de riluzole ne sont pas modifiés après administration répétée (50 mg de riluzole deux fois par jour pendant 4 jours et demi) chez les personnes âgées (> 70 ans).
Insuffisants hépatiques : après administration orale d'une dose unique de 50 mg de riluzole, l’AUC de riluzole est multipliée environ par 1,7 chez les patients insuffisants hépatiques chroniques légers et environ par 3 chez les patients insuffisants hépatiques chroniques modérés.
Origine ethnique : une étude clinique a été conduite chez 16 adultes volontaires sains, de sexe masculin, d’origine japonaise et caucasienne afin d’évaluer la pharmacocinétique du riluzole et de son métabolite Nhydroxyriluzole après administration orale bi-quotidienne répétée pendant 8 jours.
Le groupe d’origine japonaise a montré une plus faible exposition au riluzole (Cmax 0,85 [IC 90% 0,68-1,08] et une AUC inf 0,88 [IC 90% 0,69-1,13]) que le groupe caucasien et une exposition similaire au métabolite. La signification clinique de ces résultats demeure inconnue.
Sexe : une étude de bioéquivalence a été menée pour comparer TEGLUTIK (suspension buvable) et RILUTEK (comprimés). Les résultats ont montré une bioéquivalence des deux formulations chez les sujets de sexe féminin, et une exposition supérieure chez les sujets de sexe masculin en termes de Cmax et d'ASC du riluzole.
Aucun impact clinique significatif n'est cependant attendu.
5.3. Données de sécurité préclinique
Aucun potentiel carcinogène n’a été démontré chez le rat et la souris avec le riluzole.
Les études standards de génotoxicité réalisées avec le riluzole ont été négatives. Deux études in vitro réalisées avec le principal métabolite actif du riluzole ont donné des résultats positifs. Sept autres études plus approfondies réalisées in vitro ou in vivo n’ont pas mis en évidence de potentiel génotoxique pour ce métabolite.
Sur la base de ces données, et en tenant compte des études négatives sur la carcinogénicité du riluzole chez la souris et le rat, l’effet génotoxique de ce métabolite est considéré comme dénué de signification clinique chez l’homme.
Des réductions des paramètres de la lignée rouge et/ou des altérations des paramètres biologiques hépatiques ont été notées de façon inconstante dans les études de toxicité subaiguë et chronique effectuées chez le rat et le singe. Chez le chien, une anémie hémolytique a été observée.
Dans une étude de toxicité conduite chez le rat, une absence de corps jaune ovarien a été notée avec une incidence plus grande dans le groupe traité que dans le groupe témoin. Cette observation isolée n’a été enregistrée dans aucune autre étude ou espèce.
Ces données ont toutes été observées pour des doses 2 à 10 fois supérieures à la dose thérapeutique de 100 mg/jour.
Chez la rate gestante, un passage du 14C-riluzole à travers la barrière placentaire vers le foetus a été mis en évidence. Chez le rat, le riluzole entraîne une diminution du taux de grossesses et du nombre d'implantations pour des niveaux d’exposition systémique au moins 2 fois supérieurs à l’exposition systémique à la dose thérapeutique chez l’homme. Aucune malformation n’a été observée au cours des études de reproduction chez l’animal.
Chez la rate allaitante, le 14C-riluzole passe dans le lait maternel.
3 ans
Après la première ouverture : 15 jours, sans précautions particulières de conservation.
6.4. Précautions particulières de conservation
Pas de précautions particulières de conservation.
Voir rubrique 6.3 pour les conditions de conservation après la première ouverture du médicament.
6.5. Nature et contenu de l'emballage extérieur
Flacon de verre ambré muni d'un adaptateur en PEBD pour seringue, et fermé par un capuchon de sécurité blanc en PEHD.
Boîte de un ou deux flacons de 250 ml de riluzole à la concentration de 5 mg/ml en suspension buvable.
Boîte de un flacon de 300 ml de riluzole à la concentration de 5 mg/ml en suspension buvable.
Le flacon est fourni avec une seringue graduée en plastique pour administration orale. Le corps de la seringue est gradué en millilitres jusqu'à 10 ml.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
TEGLUTIK suspension buvable peut être administrée par voie orale ou également par sonde de gastrostomie.
Administration par voie orale
La suspension doit être agitée délicatement à la main pendant au moins 30 secondes en inversant le flacon de 180° ; vérifier ensuite visuellement l'homogénéité.
Ouvrir le flacon, connecter la seringue d'administration à l'adaptateur pour seringue du flacon ; retourner le flacon et le maintenir inversé ; aspirer lentement le volume de suspension correspondant à la dose recommandée (10 ml correspondent à 50 mg de riluzole).
Après la prise de la suspension, laver soigneusement la seringue avec de l'eau du robinet.
Administration par sonde de gastrostomie
TEGLUTIK suspension buvable peut être administrée par sonde de gastrostomie.
La compatibilité a été testée avec des tubes de silicone ou de polyuréthane avec de diamètres de 14Fr à 20Fr.
Il est recommandé de suivre les instructions ci-dessous :
Assurez-vous que la sonde de gastrostomie n’est pas obstruée avant l’administration.
1. Rincer la sonde de gastronomie avec 30 ml d’eau
2. Administrer la dose requise de TEGLUTIK suspension buvable avec une seringue graduée
3. Rincer la sonde de gastrostomie avec 30 ml d’eau
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
C/SAN RAFAEL
3, POL. IND. ALCOBENDAS
28108 ALCOBENDAS (MADRID)
ESPAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 275 660 1 1: 300 ml de suspension en flacon de verre avec seringue graduée (10 ml) pour administration orale (PEHD) et adaptateur (PEBD).
· 34009 300 731 7 9 : 1 flacon de 250 ml de suspension en flacon de verre avec seringue graduée (10 ml) pour administration orale (PEHD) et adaptateur (PEBD).
· 34009 300 731 8 6 : 2 flacons de 250 ml de suspension en flacon de verre avec seringue graduée (10 ml) pour administration orale (PEHD) et adaptateur (PEBD).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicaments soumis à prescription restreinte
Médicament nécessitant une surveillance particulière pendant le traitement.
Médicament soumis à prescription initiale annuelle réservée aux spécialistes en neurologie.
Renouvellement non restreint.
ANSM - Mis à jour le : 19/02/2026
TEGLUTIK 5 mg/ml, suspension buvable
riluzole
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que TEGLUTIK 5 mg/ml, suspension buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre TEGLUTIK 5 mg/ml, suspension buvable?
3. Comment prendre TEGLUTIK 5 mg/ml, suspension buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TEGLUTIK 5 mg/ml, suspension buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TEGLUTIK 5 mg/ml, suspension buvable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : autres médicaments du système nerveux, code ATC : N07XX02.
TEGLUTIK est un médicament qui agit sur le système nerveux.
Dans quel cas TEGLUTIK est-il utilisé :
TEGLUTIK est prescrit chez des patients avec une sclérose latérale amyotrophique (SLA).
La SLA est une forme de maladie du motoneurone qui affecte les cellules nerveuses responsables de l'envoi d'instructions aux muscles, ce qui entraîne une faiblesse musculaire, une atrophie musculaire et une paralysie.
La destruction des cellules nerveuses dans les maladies du motoneurone peut être provoquée par une quantité trop importante de glutamate (un messager chimique) dans le cerveau et la moelle épinière. TEGLUTIK bloque la libération de glutamate, ce qui peut contribuer à empêcher la destruction des cellules nerveuses.
Veuillez consulter votre médecin pour des informations complémentaires sur la SLA et les raisons pour lesquelles ce médicament vous a été prescrit.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE TEGLUTIK 5 mg/ml, suspension buvable ?
· si vous êtes allergique (hypersensible) au riluzole ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous avez une maladie du foie ou des taux sanguins augmentés de certaines enzymes hépatiques (transaminases),
· si vous êtes enceinte ou que vous allaitez.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre TEGLUTIK.
· Si vous avez une maladie du foie : jaunissement de la peau ou du blanc des yeux (jaunisse), démangeaisons généralisées, nausées, vomissements,
· Si vous avez une maladie des reins,
· Si vous avez de la fièvre, ceci peut être dû à une diminution du nombre des globules blancs qui peut augmenter le risque d’infections.
Si une des situations citées ci-dessus s’applique à votre cas ou si vous avez des doutes, prévenez votre médecin qui décidera de ce qu’il faut faire.
Enfants et adolescents
Si vous êtes âgé de moins de 18 ans, l’utilisation de TEGLUTIK n’est pas recommandée car il n’y a pas d’information disponible dans cette population.
Autres médicaments et TEGLUTIK
Informez votre médecin si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
TEGLUTIK avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Vous ne devez pas prendre TEGLUTIK si vous êtes enceinte ou pensez l'être ou si vous allaitez.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Ne conduisez pas ou n’utilisez pas de machines si vous avez des vertiges ou si vous vous sentez étourdis après avoir pris TEGLUTIK.
TEGLUTIK contient du sorbitol liquide (E420) et du sodium.
Ce médicament contient 4000 mg de sorbitol (E420) dans 10 ml de suspension buvable.
Le sorbitol est une source de fructose. Si votre médecin vous a informé que vous présentiez une intolérance à certains sucres ou si vous avez été diagnostiqué avec une intolérance héréditaire au fructose (IHF), un trouble génétique rare caractérisé par l'incapacité à décomposer le fructose, parlez-en à votre médecin avant que vous ne preniez ou ne receviez ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) par dose de 10 ml de suspension buvable, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE TEGLUTIK 5 mg/ml, suspension buvable ?
La suspension buvable peut être administrée par voie orale ou également par sonde de gastrostomie.
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
La dose recommandée est de 100 mg/j (50 mg toutes les 12 heures).
10 ml de suspension buvable contient 50 mg de riluzole, la dose doit être prise toutes les 12 heures, chaque jour à la même heure (par exemple, matin et soir). La suspension s'administre au moyen d'une seringue graduée.
La suspension buvable doit être agitée délicatement à la main pendant au moins 30 secondes en retournant plusieurs fois le flacon pour bien mélanger la suspension de TEGLUTIK. Il ne doit plus y avoir de surnageant clair, ni de particules au fond du flacon.
Mode d’administration
Administration par voie orale :
|
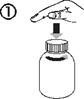
Ouverture du flacon : Pousser sur le bouchon et le tourner en sens antihoraire (Figure 1) |
|
|
|
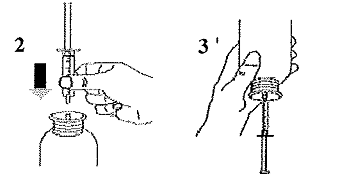
Prendre la seringue, retirer le capuchon et insérer la seringue dans l'ouverture de l'adaptateur (Figure 2). Retourner le flacon, ouverture vers le bas (Figure 3). |
|
|
|
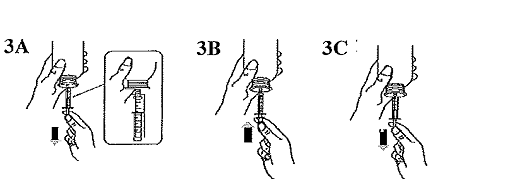
Remplir la seringue avec un petit volume de suspension en tirant sur le piston vers le bas (Figure 3A), puis repousser le piston vers le haut pour éliminer les éventuelles bulles (Figure 3B). Tirer le piston vers le bas jusqu'à la graduation qui correspond au volume en millilitres (ml) prescrit par le médecin (Figure 3C). |
|
|
|
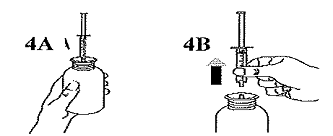
Redresser le flacon (Figure 4A). Sortir la seringue hors de l'adaptateur (Figure 4B). |
|
|
|
Ingérer l'ensemble du contenu de la seringue. Refermer le flacon avec le bouchon vissable en plastique. Laver la seringue exclusivement avec de l'eau, puis la remonter après avoir séché son capuchon (Figure 5). |
|
|
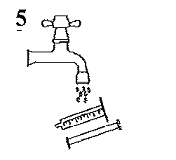
Administration par sonde de gastrostomie :
Assurez-vous que la sonde de gastrostomie n’est pas obstruée avant l’administration.
1. Rincer la sonde de gastrostomie avec 30 ml d’eau
2. Administrer la dose requise de TEGLUTIK avec une seringue graduée
3. Rincer la sonde de gastrostomie avec 30 ml d’eau
Si vous avez pris plus de TEGLUTIK que vous n’auriez dû :
Si vous avez pris un volume trop important de la suspension, contactez immédiatement votre médecin ou le service d'urgence de l'hôpital le plus proche.
Si vous oubliez de prendre TEGLUTIK :
Si vous oubliez de prendre une dose, prenez la dose suivante au moment habituel.
Ne prenez pas de dose double pour compenser la dose que vous avez oubliée de prendre.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Remarque importante
Contactez immédiatement votre médecin :
· Si vous avez de la fièvre (augmentation de la température corporelle) : TEGLUTIK peut provoquer une diminution du nombre des globules blancs. Votre médecin pourra demander un examen de sang pour vérifier le nombre de globules blancs, des cellules importantes pour combattre les infections.
· Si vous manifestez l’un des symptômes suivants : jaunissement de la peau ou du blanc des yeux (jaunisse), démangeaisons généralisées, nausées, vomissements. Ces signes peuvent indiquer une maladie du foie (hépatite). Votre médecin peut devoir demander des examens sanguins réguliers pendant le traitement avec TEGLUTIK afin d'éviter l’apparition de ces problèmes.
· Si vous présentez de la toux ou des difficultés respiratoires, cela peut indiquer une maladie pulmonaire (appelée pneumonie interstitielle).
Autres effets indésirables
Effets indésirables très fréquents (pouvant affecter plus de 1 personne sur 10) :
· fatigue
· nausées
· augmentation des taux sanguins de certaines enzymes du foie (transaminases).
Effets indésirables fréquents (pouvant affecter jusqu’à 1 personne sur 10) :
· étourdissement
· sensations d’engourdissement ou de picotement de la bouche
· vomissements,
· somnolence
· accélération du rythme cardiaque
· diarrhée
· maux de tête
· douleur abdominale,
· douleurs.
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) :
· anémie
· réactions allergiques,
· inflammation du pancréas (pancréatite)
Inconnus :
· Éruption cutanée
Comme le riluzole en suspension buvable est absorbé plus rapidement que le riluzole en comprimés, on ne peut pas exclure une légère augmentation de la fatigue, des vertiges, de la diarrhée et des taux des transaminases.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER TEGLUTIK 5 mg/ml, suspension buvable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage et sur le flacon, après l’abréviation utilisée pour la mention « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de conditions particulières de conservation.
Après ouverture : à utiliser dans les 15 jours.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Riluzole ............................................................................................................................ 5 mg
Pour 1 ml de suspension buvable.
· Les autres composants sont :
Sorbitol liquide (E420), silicate d'aluminium et de magnésium, gomme xanthane (E415), saccharine sodique (E954), émulsion de siméthicone 30 %, laurylsulfate de sodium, éther cétostéarylique de macrogol, eau.
Qu’est-ce que TEGLUTIK et contenu de l’emballage extérieur
Ce médicament se présente sous la forme d'une suspension buvable opaque, homogène et légèrement brune après avoir été doucement agitée.
TEGLUTIK est fourni en flacon de 250 ml et de 300 ml avec une seringue graduée en plastique pour administration orale.
Les conditionnements sont les suivants :
· Boîte contenant un ou deux flacons de 250 ml de riluzole à la concentration de 5 mg/ml en suspension buvable.
· Boîte de un flacon de 300 ml de riluzole à la concentration de 5 mg/ml en suspension buvable.
Le corps de la seringue est gradué en millilitres jusqu'à 10 ml.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
C/SAN RAFAEL
3, POL. IND. ALCOBENDAS
28108 ALCOBENDAS (MADRID)
ESPAGNE
Exploitant de l’autorisation de mise sur le marché
EFFIK
Batiment « Le Newton »
11 rue Jeanne Braconnier
92360 Meudon
C/SAN RAFAEL
3, POL. IND. ALCOBENDAS
28108 ALCOBENDAS (MADRID)
ESPAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).