Dernière mise à jour le 01/06/2026
DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours
Indications thérapeutiques
Ce médicament contient de la triptoréline. La triptoréline appartient à un groupe de médicaments connus sous le nom d’analogues de l’hormone entraînant la libération de gonadotrophines (GnRH). L’une de ses actions est de diminuer la production d’hormones sexuelles dans le corps.
Ce médicament est un analogue d’une hormone naturelle.
Il est utilisé :
Chez l’homme adulte :
· dans le traitement du cancer de la prostate localisé à haut risque ou localement avancé, en association à la radiothérapie.
Chez l’enfant :
· dans le traitement de la puberté qui survient prématurément, c’est-à-dire avant 8 ans chez les filles et 10 ans chez les garçons (puberté précoce centrale).
Chez la femme :
· dans le traitement de l’endométriose.
· dans le traitement de certaines stérilités. Ce médicament est alors généralement associé avec d’autres hormones (appelées gonadotrophines) au cours des procédures de fécondation in vitro (FIVETE).
· dans le traitement pré-opératoire de certains fibromes utérins.
· dans le traitement du cancer du sein hormonosensible au stade précoce chez les femmes non ménopausées ayant reçu une chimiothérapie. DECAPEPTYL L.P. 3 mg est utilisé en association avec des médicaments hormonaux. Vous devrez également prendre :
o un médicament appelé tamoxifène - on vous demandera de prendre ce médicament si vous êtes à haut risque de récidive du cancer
ou
o un médicament «inhibiteur de l'aromatase» tel que l’exémestane. Vous serez traitée avec DECAPEPTYL L.P. 3 mg pendant au moins 6 à 8 semaines avant de commencer à prendre ce médicament.
N'oubliez pas de lire la notice du médicament que vous prenez en association avec DECAPEPTYL L.P. 3 mg.
Présentations
> 1 flacon(s) en verre de poudre - 1 ampoule(s) en verre de 2 ml de solvant avec 1 seringue(s) avec 2 aiguille(s)
Code CIP : 339 437-6 ou 34009 339 437 6 9
Déclaration de commercialisation : 19/03/1996
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 85,97 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 86,99 €
- Taux de remboursement :65%
- Endométriose
- infécondité féminine
- fibrome utérin
- puberté précoce
- cancer de la prostate, dans certains cas. ; JOURNAL OFFICIEL ; 25/08/06
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Insuffisant | Avis du 19/09/2018 | Extension d'indication | le service médical rendu par DECAPEPTYL LP 3 mg est insuffisant pour justifier d’une prise en charge par la solidarité nationale dans l’indication « Traitement adjuvant, en association avec le tamoxifène ou un inhibiteur de l’aromatase, du cancer du sein hormonosensible à un stade précoce chez des femmes à haut risque de récidive, confirmées comme non ménopausées à l’issue d’une chimiothérapie. ». |
| Important | Avis du 25/07/2018 | Extension d'indication | Le service médical rendu par les spécialités DECAPEPTYL LP 3 mg, 11,25 mg et 22,5 mg est important, en association avec la radiothérapie dans le traitement du cancer de la prostate localisé à haut risque. |
| Important | Avis du 16/03/2016 | Renouvellement d'inscription (CT) | Le service médical rendu par les spécialités DECAPEPTYL reste important dans les indications de leur AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 25/07/2018 | Extension d'indication | Les spécialités DECAPEPTYL LP 3 mg, 11,25 mg et 22,5 mg, en association avec la radiothérapie, n’apportent pas d’amélioration du service médical rendu (ASMR V) dans la prise charge du cancer de la prostate localisé à haut risque. Pour rappel, cette prise en charge comprend les autres analogues de la GnRH mentionnés dans la rubrique « 06. Comparateurs cliniquement pertinents » de cet avis. |
| V (Inexistant) | Avis du 12/04/2006 | Extension d'indication | DECAPEPTYL LP n’apporte pas d’amélioration du service médical rendu (ASMR de niveau V) par rapport aux autres spécialités à base d’un analogue de la GnRH indiquées dans le traitement du cancer de la prostate à un stade localement avancé. |
ANSM - Mis à jour le : 11/06/2025
DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Triptoréline......................................................................................................................... 3,00 mg*
(sous forme de pamoate de triptoréline)
Pour une unité de prise
* Compte tenu des caractéristiques de la forme pharmaceutique, chaque flacon contient une quantité de pamoate de triptoréline correspondant à 4,3 mg de triptoréline.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour suspension injectable (I.M.) à libération prolongée.
4.1. Indications thérapeutiques
Traitement du cancer de la prostate localement avancé ou métastatique.
Traitement du cancer de la prostate localisé à haut risque ou localement avancé, en association à la radiothérapie. Voir rubrique 5.1.
Un effet favorable du médicament est d'autant plus net et plus fréquent que le patient n'a pas reçu auparavant un autre traitement hormonal.
· Puberté précoce centrale (avant 8 ans chez la fille, avant 10 ans chez le garçon).
· Endométriose à localisation génitale et extragénitale (du stade I au stade IV)
La durée du traitement est limitée à 6 mois (voir rubrique 4.8). Il n'est pas souhaitable d'entreprendre une seconde cure par la triptoréline ou par un autre analogue de la GnRH.
· Infécondité féminine
Traitement complémentaire, en association avec les gonadotrophines (hMG, FSH, hCG), au cours d'une induction de l'ovulation en vue d'une fécondation in vitro suivie d'un transfert d'embryon (FIVETE).
· Traitement pré-opératoire des fibromes utérins :
o associés à une anémie (avec un taux d’hémoglobine inférieur ou égal à 8 g/dl),
o dans le cas où une réduction de la taille du fibrome est nécessaire pour faciliter ou modifier la technique opératoire : chirurgie endoscopique, chirurgie transvaginale.
La durée du traitement est limitée à 3 mois.
· Cancer du sein
Traitement adjuvant, en association avec le tamoxifène ou un inhibiteur de l'aromatase, du cancer du sein hormonosensible à un stade précoce chez des femmes à haut risque de récidive, confirmées comme non ménopausées à l’issue d’une chimiothérapie (voir rubriques 4.3, 4.4, 4.8 et 5.1).
4.2. Posologie et mode d'administration
· Cancer de la prostate :
Deux schémas thérapeutiques peuvent être utilisés :
Une injection sous-cutanée quotidienne de DECAPEPTYL 0,1 mg à libération immédiate pendant sept jours, puis une injection intramusculaire de DECAPEPTYL L.P. 3 mg le 8ème jour ; cette injection est répétée toutes les 4 semaines, ou d'emblée, une injection intramusculaire de DECAPEPTYL L.P. 3 mg à libération prolongée qui sera renouvelée toutes les 4 semaines.
Durée du traitement :
Dans le traitement du cancer de la prostate hormonodépendant localisé à haut risque ou localement avancé, en traitement concomitant et adjuvant à la radiothérapie, les données cliniques ont montré que la radiothérapie suivie par un traitement de privation androgénique longue était préférable à une radiothérapie suivie par une privation androgénique courte. Voir rubrique 5.1.
La durée du traitement de privation androgénique recommandée par les recommandations cliniques pour les patients avec un cancer de la prostate localisé à haut risque, ou localement avancé, recevant une radiothérapie est de 2 à 3 ans.
Chez les patients atteints d’un cancer de la prostate métastatique résistant à la castration, non castrés chirurgicalement, traités par un agoniste de la GnRH, comme la triptoréline, et éligibles à un traitement par l’acétate d’abiratérone, un inhibiteur de la biosynthèse des androgènes ou l’enzalutamide, un inhibiteur de la voie de signalisation des récepteurs aux androgènes, le traitement par un agoniste de la GnRH doit être poursuivi.
· Puberté précoce centrale :
Le traitement doit être supervisé par un endocrinologue pédiatre, un pédiatre ou un endocrinologue spécialisé dans le traitement de la puberté précoce centrale.
Enfants de moins de 20 kg : injection intramusculaire toutes les 4 semaines (28 jours) d’une ½ (demie) dose (soit la moitié du volume de la suspension reconstituée).
Enfants de 20 à 30 kg : injection intramusculaire toutes les 4 semaines (28 jours) de 2/3 (deux tiers) de la dose (soit les 2/3 du volume de la suspension reconstituée).
Enfants de plus de 30 kg : injection intramusculaire toutes les 4 semaines (28 jours) de toute la dose (soit tout le volume de la suspension reconstituée).
Le traitement doit être arrêté vers l'âge physiologique de la puberté chez les garçons et les filles et il est recommandé de ne pas poursuivre le traitement chez les filles ayant un âge osseux supérieur à 12-13 ans. Chez les garçons, il existe peu de données disponibles concernant l’âge osseux optimal pour arrêter le traitement. Toutefois, il est recommandé d’arrêter le traitement chez les garçons ayant un âge osseux de 13-14 ans.
· Endométriose :
Une injection intramusculaire de DECAPEPTYL L.P. 3 mg renouvelée toutes les 4 semaines.
Le traitement doit être débuté dans les 5 premiers jours du cycle.
Durée du traitement : elle est fonction de la gravité initiale de l'endométriose et de l'évolution sous traitement de ses manifestations cliniques (fonctionnelles et anatomiques). La durée du traitement est limitée à 6 mois (voir rubrique 4.8). Il n'est pas souhaitable d'entreprendre une deuxième cure par la triptoréline ou par les autres analogues de la GnRH.
· Infécondité féminine :
Le schéma thérapeutique habituel est basé sur l'injection intramusculaire d'un flacon de DECAPEPTYL L.P. 3 mg à partir du 2ème jour du cycle.
L'association aux gonadotrophines débute après l'obtention de la désensibilisation hypophysaire (taux des œstrogènes plasmatiques inférieur à 50 pg/ml), en général une quinzaine de jours après l'injection du DECAPEPTYL L.P 3 mg.
· Traitement pré-opératoire des fibromes utérins :
Une injection intramusculaire de DECAPEPTYL L.P. 3 mg renouvelée toutes les 4 semaines.
Voie Intramusculaire uniquement.
Le traitement doit débuter dans les 5 premiers jours du cycle.
La durée du traitement est limitée à 3 mois.
· Cancer du sein
Une injection intramusculaire renouvelée toutes les 4 semaines en association avec le tamoxifène ou un inhibiteur de l'aromatase.
Le traitement par la triptoréline doit être initié à l’issue de la chimiothérapie, une fois que l’absence de ménopause a été confirmée (voir rubrique 4.4).
Le traitement par la triptoréline doit être initié au moins 6 à 8 semaines avant de débuter le traitement par inhibiteur de l'aromatase. Au minimum deux injections de triptoréline (avec un intervalle de 4 semaines entre les injections) doivent être administrées avant le début du traitement par inhibiteur de l'aromatase.
Pendant le traitement par un inhibiteur de l'aromatase, le traitement par la triptoréline ne doit pas être interrompu afin d’éviter une augmentation rebond des œstrogènes circulants chez les femmes non ménopausées.
La durée recommandée du traitement adjuvant en association avec d'autres hormonothérapies peut aller jusqu’à 5 ans.
Voir section « Posologie » ci-dessus.
Comme DECAPEPTYL L.P. 3 mg est une suspension de microparticules, une injection intravasculaire doit être strictement évitée.
N.B. Il est important que l'injection de la forme à libération prolongée soit pratiquée rigoureusement selon les instructions de la notice. Toute injection défectueuse conduisant à la perte d'une quantité de la suspension supérieure à celle qui reste normalement dans le dispositif utilisé pour l'injection, doit être signalée.
· Hypersensibilité à la GnRH, aux analogues de la GnRH, ou à l’un des excipients mentionnés à la rubrique 6.1 (voir rubrique 4.8).
· Grossesse et allaitement.
· Dans le traitement du cancer du sein chez la femme non ménopausée : initiation du traitement par inhibiteur de l'aromatase avant l’obtention d’une suppression de la fonction ovarienne suffisante par la triptoréline (voir rubriques 4.2 et 4.4).
4.4. Mises en garde spéciales et précautions d'emploi
Rarement, le traitement par les analogues de la GnRH peut révéler la présence jusque là inconnue d’un adénome hypophysaire gonadotrope. Ces patients peuvent présenter une apoplexie pituitaire se caractérisant par l’apparition d’une céphalée soudaine, de vomissements, de troubles visuels et d’une ophtalmoplégie.
Il y a un risque accru de survenue d’une dépression (potentiellement sévère) chez les patients traités par agonistes de la GnRH, comme la triptoréline. Les patients doivent être informés en conséquence et traités de façon appropriée si des symptômes apparaissent. Les patients qui souffrent de dépression doivent faire l’objet d’un suivi adapté pendant le traitement.
Des convulsions ont été rapportées avec les analogues de la GnRH, en particulier chez les femmes et les enfants. Certains de ces patients présentaient des facteurs de risque de convulsions (tels que des antécédents d'épilepsie, des tumeurs intracrâniennes ou un traitement concomitant avec des médicaments connus pour présenter un risque de crises convulsives). Des convulsions ont également été rapportées chez des patients ne présentant pas de tels facteurs de risque.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu’il est essentiellement « sans sodium ».
La prudence est requise chez les patients traités par des anticoagulants, en raison du risque potentiel d’hématome au site d’injection.
Chez l’homme
Au début du traitement, la triptoréline comme les autres agonistes de la GnRH provoque une augmentation transitoire du taux de testostérone sérique. Cela peut conduire à des cas isolés d’aggravation transitoire des signes et symptômes du cancer de la prostate lors des premières semaines de traitement. Lors de la phase initiale du traitement, l’administration additionnelle d’un anti-androgène adapté devra être considérée afin de neutraliser l’augmentation initiale du taux sérique de testostérone et l’aggravation des symptômes cliniques.
Chez un nombre réduit de patients peuvent survenir une exacerbation de la tumeur avec une aggravation transitoire des signes et symptômes du cancer de la prostate (poussée tumorale) et une augmentation temporaire des douleurs liées au cancer (douleurs métastatiques), qui peuvent être traitées symptomatiquement.
Comme avec les autres agonistes de la GnRH, des cas isolés de compression médullaire ou d’obstruction de l’urètre ont été observés. En cas de compression médullaire ou d’insuffisance rénale, un traitement standard de ces complications devra être institué et dans les cas extrêmes une orchidectomie d’urgence envisagée (castration chirurgicale). Un suivi attentif est nécessaire lors des premières semaines de traitement, en particulier chez les patients souffrant de métastases vertébrales, à haut risque de compression médullaire, et/ou d’obstruction des voies urinaires. Pour la même raison la mise en route du traitement chez les sujets présentant des signes prémonitoires de compression médullaire doit être soigneusement pesée.
Après castration chirurgicale, la triptoréline ne provoque pas de diminution supplémentaire des taux de testostérone.
L’inhibition prolongée de la sécrétion androgénique qu’elle soit induite par orchidectomie bilatérale ou par administration d’analogue de la GnRH est associée à un risque élevé de perte osseuse et peut conduire à une ostéoporose et à un risque accru de fracture osseuse.
Le traitement par suppression androgénique peut provoquer un allongement de l’intervalle QT.
Chez les patients ayant des antécédents ou présentant des facteurs de risque d’allongement de l’intervalle QT et chez les patients traités par un médicament susceptible de prolonger l’intervalle QT (voir rubrique 4.5), le médecin devra évaluer le rapport bénéfice/risque, y compris le risque de torsades de pointe, avant l’initiation d’un traitement par DECAPEPTYL L.P. 3 mg.
De plus, des données épidémiologiques ont mis en évidence que ces patients pouvaient être sujets à des changements métaboliques (par exemple une intolérance au glucose, une stéatose hépatique), ou un risque plus élevé de maladie cardiovasculaire pendant le traitement inhibant la sécrétion androgénique. Toutefois, les données prospectives ne confirment pas le lien entre le traitement par analogue de la GnRH et l’augmentation de la mortalité cardiovasculaire. Les patients qui ont un risque élevé de maladies cardiovasculaires ou métaboliques doivent faire l’objet d’une évaluation attentive avant d’instaurer le traitement et d’un suivi adapté pendant le traitement inhibant la sécrétion androgénique.
Du fait de la suppression androgénique, le traitement par les analogues de la GnRH peut augmenter le risque d’anémie. Ce risque doit être évalué chez les patients traités et pris en charge de façon appropriée.
L’administration de triptoréline aux doses thérapeutiques conduit à une suppression du système gonadotrope hypophysaire. Un retour à la normale est généralement obtenu après l’interruption du traitement. Des tests diagnostiques de la fonction gonadotrope hypophysaire conduits durant le traitement et après l’interruption de la thérapie avec un analogue de la GnRH peuvent donc être erronés.
Une élévation transitoire des phosphatases acides en début de traitement peut être observée.
Chez la femme
Il est indispensable de vérifier l’absence de grossesse avant toute prescription de DECAPEPTYL L.P. 3 mg.
L’utilisation des agonistes de la GnRH est susceptible de provoquer une diminution de la densité minérale osseuse. Cette réduction est d’environ 1% par mois, au cours d’un traitement de 6 mois. Une réduction de 10% de la densité minérale osseuse est corrélée à une multiplication du risque de fracture par 2 à 3.
Aucune donnée spécifique n’est disponible chez les patientes qui ont déjà une ostéoporose ou qui ont des facteurs de risque d’ostéoporose (par exemple : alcoolisme chronique, tabagisme, traitements à long terme avec des médicaments qui réduisent la densité minérale osseuse, comme par exemple les antiépileptiques ou les corticoïdes, antécédents familiaux d’ostéoporose, malnutrition liée par exemple à une anorexie mentale). Comme la réduction de la densité minérale osseuse est susceptible d'être plus néfaste chez ces patientes, l’instauration d’un traitement par la triptoréline devra être soigneusement évaluée au cas par cas en s’assurant que le bénéfice attendu est supérieur au risque. Des mesures supplémentaires visant à limiter la perte de densité minérale osseuse pourront être envisagées.
· Infécondité féminine
Il est indispensable de vérifier l’absence de grossesse avant toute prescription de DECAPEPTYL L.P. 3 mg.
L'augmentation du recrutement folliculaire induit par l'injection de triptoréline, lorsqu'elle est associée aux gonadotrophines, peut être importante chez certaines patientes prédisposées et en particulier en cas d'ovaires polykystiques. Comme avec les autres analogues de la GnRH des cas de syndrome d’hyperstimulation ovarienne ont été rapportés lors du traitement par les gonadotrophines associé à la triptoréline.
La réponse ovarienne à l'association triptoréline-gonadotrophines peut varier avec les mêmes doses d'une patiente à l'autre et, dans certains cas, d'un cycle à l'autre pour une même patiente.
Chez les insuffisants rénaux ou les insuffisants hépatiques, la triptoréline a une demi-vie terminale de 7 à 8 heures au lieu de 3 à 5 heures chez les sujets sains. Malgré cette exposition prolongée, la triptoréline ne devrait pas être présente dans la circulation au moment du transfert d'embryon.
L'induction de l'ovulation ne doit être réalisée que sous étroite surveillance médicale avec contrôles biologiques et cliniques stricts et réguliers : dosages des œstrogènes plasmatiques, échographies (voir rubrique 4.8).
En cas de réponse ovarienne excessive, il est recommandé d'interrompre le cycle de stimulation en cessant les injections de gonadotrophines.
· Endométriose et traitement pré-opératoire des fibromes utérins
Chez les patientes avec une endométriose traitée par les analogues de la GnRH, il a été montré que l’ajout d’une ABT (comportant un œstrogène et un progestatif) réduisait la perte de densité minérale osseuse et les symptômes vasomoteurs (voir rubrique 4.2).
L’utilisation de DECAPEPTYL L.P. 3 mg à la posologie recommandée entraîne une aménorrhée hypogonadotrophique constante.
La survenue de métrorragies au cours du traitement en dehors du premier mois est anormale : elle doit conduire à la vérification du taux d’œstradiol plasmatique et s’il est inférieur à 50 pg/ml, il faut rechercher d’éventuelles lésions organiques associées.
Après l’arrêt du traitement, la fonction ovarienne reprend et l’ovulation survient environ 2 mois après la dernière injection.
Une méthode de contraception non-hormonale devra être utilisée tout au long du traitement et jusqu’à 1 mois après la dernière injection.
Au cours du traitement des fibromes utérins, il est recommandé de contrôler régulièrement la taille du fibrome. Quelques cas de saignements ont été rapportés chez des patientes avec des fibromes sous-muqueux. Les saignements survenaient généralement 6 à 10 semaines après le début du traitement.
La patiente devra être informée qu’elle doit consulter son médecin en cas de persistance des règles.
· Cancer du sein
Afin d'assurer une suppression de la fonction ovarienne suffisante chez les femmes non ménopausées, le traitement par la triptoréline doit être administré pendant au moins 6 à 8 semaines avant le début d'un traitement par inhibiteur de l'aromatase, et les injections de triptoréline doivent être administrées comme prévu toutes les 4 semaines et sans interruption pendant le traitement par inhibiteur de l'aromatase.
Chez les femmes non ménopausées au moment du diagnostic et devenant aménorrhéiques après la chimiothérapie, la production d'œstrogènes par les ovaires peut perdurer ou non. Après la chimiothérapie et avant le début du traitement par la triptoréline, indépendamment du statut menstruel, le statut non ménopausique devrait être confirmé, par des concentrations sanguines d'œstradiol et d'hormone folliculo-stimulante (FSH) conformes aux normes des femmes non ménopausées, afin d'éviter un traitement inutile par la triptoréline en cas de ménopause induite par la chimiothérapie. Après le début du traitement par la triptoréline, il est important de confirmer une suppression ovarienne adéquate (ménopause induite par l'analogue de la GnRH) par une évaluation régulière du taux de FSH circulant et d’œstradiol pour s’assurer d’un état post-ménopausique réel, si un traitement par inhibiteur de l'aromatase est envisagé dans cette sous-population de femmes, conformément aux recommandations pour la pratique clinique en vigueur.
Par conséquent, la suppression de la fonction ovarienne doit être confirmée par des taux sanguins bas de FSH et d’œstradiol avant de débuter le traitement par inhibiteur de l’aromatase et les dosages doivent être répétés tous les 3 mois pendant la durée de l’association de la triptoreline avec un inhibiteur de l’aromatase.
Ceci a pour objectif d’éviter une augmentation rebond des taux d’œstrogènes circulants induite par l'inhibiteur de l'aromatase avec des répercutions sur le cancer du sein. Il est à noter que les taux de FSH circulants sont abaissés en réponse à la suppression de la fonction ovarienne induite par l’inhibition de la fonction gonadotrope par l'analogue de la GnRH (ménopause induite), contrairement à une ménopause naturelle où les taux de FSH sont élevés.
La triptoréline, lorsqu'elle est utilisée comme traitement adjuvant en association avec le tamoxifène ou un inhibiteur de l'aromatase, est associée à un risque élevé d'ostéoporose. Une ostéoporose a été rapportée plus fréquemment lors de l'utilisation de la triptoréline en association avec un inhibiteur de l'aromatase qu’en association avec le tamoxifène (39% versus 25%).
La densité minérale osseuse doit être évaluée avant le début du traitement par la triptoréline, en particulier chez les femmes qui présentent de multiples facteurs de risque d'ostéoporose. Ces patientes doivent être étroitement surveillées et le traitement ou la prophylaxie de l'ostéoporose doivent être initiés lorsque cela est approprié.
Le traitement des femmes non ménopausées atteintes d'un cancer du sein hormonosensible à un stade précoce, avec la triptoréline en association avec le tamoxifène ou un inhibiteur de l'aromatase doit faire suite à une évaluation individuelle attentive des risques et des bénéfices.
Les patientes qui ont arrêté le traitement par la triptoréline doivent également arrêter les inhibiteurs de l'aromatase dans le mois suivant la dernière administration de la triptoréline (formulation 28 jours).
Le risque de troubles musculo-squelettiques (dont les douleurs articulaires ou musculo-squelettiques) lorsque la triptoréline est utilisée en association avec un inhibiteur de l'aromatase ou le tamoxifène est respectivement d'environ 89% et 76%.
L'hypertension a été très fréquemment rapportée comme effet indésirable faisant l’objet d’une surveillance particulière lors de l’association de la triptoréline avec l'exémestane ou le tamoxifène (voir rubrique 4.8). Les femmes non ménopausées atteintes d'un cancer du sein recevant de la triptoréline en association avec l'exémestane ou le tamoxifène doivent faire l’objet d’un suivi régulier concernant les facteurs de risque cardiovasculaire et la pression artérielle.
L'hyperglycémie et le diabète ont été fréquemment rapportés comme effets indésirables faisant l’objet d’une surveillance particulière lors de l’association de la triptoréline avec l'exémestane ou le tamoxifène (voir rubrique 4.8). Les femmes non ménopausées atteintes d'un cancer du sein recevant de la triptoréline en association avec l'exémestane ou le tamoxifène doivent faire l'objet d'un suivi régulier concernant les facteurs de risque de diabète avec une surveillance régulière de la glycémie et un traitement antidiabétique doit être initié si nécessaire, selon les recommandations nationales.
Une dépression est survenue chez environ 50% des patientes traitées par la triptoréline en association avec le tamoxifène ou l'exémestane dans tous les groupes de traitement des études TEXT et SOFT, mais moins de 5% des patientes présentaient une dépression sévère (grade 3-4).
Les patientes doivent être informées en conséquence et traitées comme il convient en cas d’apparition de symptômes. Les patientes souffrant de dépression ou ayant des antécédents connus de dépression doivent être surveillées attentivement pendant le traitement.
Une attention particulière doit également être portée aux résumés des caractéristiques du produit de l’exémestane et du tamoxifène concernant les informations de sécurité pertinentes lorsqu’ils sont administrés en association avec la triptoréline.
La chimiothérapie peut induire une aménorrhée temporaire ou une perte permanente de la fonction ovarienne due aux dommages cytotoxiques sur le tissu gonadique. Le maintien du statut non ménopausé à l’issue de la chimiothérapie doit être confirmé conformément aux recommandations cliniques, par des taux sanguins d'œstradiol et de FSH compris dans les intervalles de référence pour les femmes non ménopausées.
Population pédiatrique
· Puberté précoce centrale
Chez les filles, il est indispensable de vérifier l’absence de grossesse avant toute prescription de triptoréline.
Le traitement des enfants avec une tumeur cérébrale évolutive doit faire l’objet d’une évaluation individuelle attentive du rapport bénéfices risques du traitement.
Les pseudo-pubertés précoces (hyperplasie ou tumeur des glandes surrénales ou des gonades) et les pubertés précoces indépendantes des gonadotrophines (testotoxicose, hyperplasie familiale des cellules de Leydig) devront être exclues.
Chez les filles, à l’initiation du traitement, la stimulation ovarienne initiale suivie par la diminution des taux d’œstrogènes induite par le traitement, peuvent conduire, au cours du premier mois, à des saignements vaginaux d’intensité légère ou modérée.
Après l’arrêt du traitement, le développement pubertaire reprend.
Les données relatives à la fertilité des patientes traitées par les analogues de la GnRH pendant l’enfance sont limitées. Chez la plupart des filles des règles régulières commencent environ 1 an après la fin du traitement.
Le traitement par les analogues de la GnRH peut entraîner une diminution de la densité minérale osseuse (DMO). Toutefois, après l'arrêt du traitement le bilan ultérieur de la masse osseuse est préservé, et le pic de croissance de la masse osseuse à la fin de l'adolescence ne semble pas être affecté par le traitement.
Une épiphysiolyse de la hanche peut se produire après l’arrêt du traitement. Il se pourrait que ce soit consécutif à l’affaiblissement du cartilage de conjugaison en raison des faibles concentrations en œstrogène pendant le traitement et à l’augmentation de la vitesse de croissance qui se produit après l’arrêt du traitement et qui faciliterait le déplacement des épiphyses.
Une hypertension intracrânienne idiopathique (pseudotumor cerebri) a été signalée chez des patients pédiatriques recevant de la triptoréline. Les patients doivent être avertis des signes et des symptômes d'hypertension intracrânienne idiopathique, notamment des céphalées sévères ou récurrentes, des troubles de la vision et des acouphènes. En cas d'hypertension intracrânienne idiopathique, l'arrêt de la triptoréline doit être envisagé.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Etant donné qu’un traitement par suppression androgénique peut provoquer un allongement de l’intervalle QT, l’utilisation concomitante de triptoréline et de médicaments connus pour allonger l’intervalle QT ou de médicaments susceptibles de provoquer des torsades de pointe tels les médicaments antiarythmiques de classe Ia (par exemple : quinidine, disopyramide) ou de classe III (par exemple : amiodarone, sotalol, dofetilide, ibutilide), la méthadone, la moxifloxacine, les antipsychotiques, etc doit être évaluée avec attention (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
La triptoréline ne doit pas être utilisée pendant la grossesse car l’utilisation des agonistes de la GnRH est associée à un risque théorique d’avortement ou d’anomalie fœtale. Avant traitement les patientes en âge de procréer doivent être examinées attentivement pour vérifier l’absence de grossesse. Des méthodes de contraception non hormonale devront être utilisées jusqu’au retour des règles.
Allaitement
La triptoréline ne doit pas être utilisée pendant l’allaitement.
Fertilité :
Quand la triptoréline est utilisée dans le cadre d’un traitement de l’infertilité, il n'existe pas de données cliniques suggérant un lien de causalité entre la triptoréline, et toute anomalie ultérieure du développement des ovocytes ou de la grossesse ou de l’issue de la grossesse.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Cependant, l’aptitude à conduire des véhicules et utiliser des machines peut être altérée en cas de survenue de sensations vertigineuses, d’une somnolence et de troubles de la vision, qui sont des effets indésirables possibles du traitement ou qui peuvent résulter de la pathologie traitée.
Tolérance générale chez l’homme (voir rubrique 4.4)
Etant donné que les patients souffrant de cancer de la prostate hormono-dépendant, localement avancé ou métastatique sont généralement âgés et ont d’autres maladies fréquemment observées dans cette population âgée, des effets indésirables ont été rapportés chez plus de 90% des patients inclus dans les essais cliniques et le lien de causalité est souvent difficile à évaluer. Comme il a été observé avec d’autres agonistes de la GnRH ou après castration chirurgicale, les effets indésirables les plus communément observés lors du traitement avec la triptoréline étaient dus à ses effets pharmacologiques attendus. Ces effets incluaient des bouffées de chaleur et une diminution de la libido.
A l’exception des réactions immuno-allergiques (rares) et des réactions au site d’injection (<5%), tous les effets indésirables sont connus pour être liés aux changements de la testostéronémie.
Les effets indésirables suivants ont été rapportés et sont considérés comme étant au moins possiblement liés au traitement par la triptoréline. La plupart de ces effets sont connus comme étant liés à une castration biochimique ou chirurgicale.
La fréquence de ces effets indésirables peut être classée comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000). Aucune fréquence ne peut être appliquée aux effets indésirables rapportés après la commercialisation. Ils sont donc rapportés avec la mention « fréquence indéterminée ».
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Fréquence indéterminée |
|
Infections et infestations |
|
|
|
Rhinopharyngite |
|
|
Affections hématologiques et du système lymphatique |
|
Anémie |
Thrombocytose |
|
|
|
Affections du système immunitaire |
|
Hypersensibilité |
|
Réaction anaphylactique |
Choc anaphylactique |
|
Troubles du métabolisme et de la nutrition |
|
|
Anorexie Diabète Goutte Hyperlipidémie Appétit augmenté |
|
|
|
Affections psychiatriques |
Diminution de la libido |
Dépression* Perte de la libido Changements d’humeur* |
Insomnie Irritabilité |
État confusionnel Baisse de l’activité Humeur euphorique |
Anxiété |
|
Affections du système nerveux |
Paresthésie du membre inférieur |
Sensation vertigineuse Céphalée |
Paresthésie |
Atteinte de la mémoire |
|
|
Affections endocriniennes |
|
|
|
|
Apoplexie hypophysaire** |
|
Affections oculaires |
|
|
Défauts visuels |
Sensation anormale dans l’œil Perturbation visuelle |
|
|
Affections de l'oreille et du labyrinthe |
|
|
Acouphènes Vertige |
|
|
|
Affections cardiaques |
|
|
Palpitations |
|
Allongement de l’intervalle QT (voir rubriques 4.4 et 4.5) |
|
Affections vasculaires |
Bouffées de chaleur |
Hypertension |
|
Hypotension |
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Dyspnée Epistaxis
|
Orthopnée |
|
|
Affections gastro-intestinales |
|
Bouche sèche Nausées |
Douleur abdominale Constipation Diarrhée Vomissement |
Distension abdominale Dysgueusie Flatulence |
|
|
Affections de la peau et du tissu sous-cutané |
Hyperhidrose |
|
Acné Alopécie Erythème Prurit Rash Urticaire |
Eruption bulleuse Purpura |
Œdème angioneurotique
|
|
Affections musculo-squelettiques et systémiques |
Douleur dorsale |
Douleur musculo-squelettique Extrémités douloureuses |
Arthralgie Douleur osseuse Crampe musculaire Faiblesse musculaire Myalgie |
Raideur articulaire Tuméfaction articulaire Raideur musculo-squelettique Ostéoarthrite |
|
|
Affections du rein et des voies urinaires |
|
|
Nycturie Rétention urinaire |
|
Incontinence urinaire |
|
Affections des organes de reproduction et du sein |
Dysérection (incluant anéjaculation, trouble de l'éjaculation) |
Douleur pelvienne |
Gynécomastie Douleur mammaire Atrophie testiculaire Douleur testiculaire |
|
|
|
Troubles généraux et anomalies au site d'administra-tion |
Asthénie |
Réaction au site d’injection (incluant érythème, inflammation et douleur) Œdème |
Léthargie Œdèmes périphériques Douleur Frissons Somnolence |
Douleur thoracique Dystasie Syndrome pseudo-grippal Fièvre |
Malaise |
|
Investigations |
|
Poids augmenté |
Alanine aminotransférase augmentée Aspartate aminotransférase augmentée Créatininémie augmentée Pression artérielle augmentée Urémie augmentée Gamma-glutamyltransférase augmentée Poids abaissé |
Phosphatase alcaline augmentée |
|
*Cette fréquence est basée sur la fréquence observée dans la classe pour tous les agonistes de la GnRH.
**Rapporté après l’administration initiale chez des patients avec un adénome hypophysaire.
La triptoréline provoque une augmentation transitoire des taux circulants de testostérone durant la première semaine après la première injection de la formulation à libération prolongée. A la suite de l’augmentation initiale des taux circulants de testostérone, un faible pourcentage de patients (≤ 5%) peut présenter une aggravation temporaire des signes et des symptômes de leur cancer de la prostate (exacerbation de la tumeur), se manifestant généralement par une augmentation des symptômes urinaires (< 2%) et des douleurs métastatiques (5%), qui peuvent être traitées de façon symptomatique. Ces symptômes sont transitoires et disparaissent généralement après 1 à 2 semaines.
Des cas isolés d’exacerbation des symptômes liés à la maladie tels qu’une obstruction de l’urètre ou une compression médullaire par des métastases peuvent survenir. C’est pourquoi les patients avec des lésions métastatiques vertébrales et/ou avec une obstruction de l’appareil urinaire doivent faire l’objet d’une surveillance attentive pendant les premières semaines de traitement (voir rubrique 4.4).
L’utilisation des agonistes de la GnRH de synthèse dans le traitement du cancer de la prostate peut être associée à une perte osseuse qui peut conduire à une ostéoporose et augmenter le risque de fracture.
Une augmentation des lymphocytes a été rapportée chez des patients traités par des analogues de la GnRH. Cette lymphocytose secondaire est apparemment liée à la castration induite par la GnRH et semble indiquer que les hormones gonadiques sont impliquées dans l’involution thymique.
Les patients traités au long cours par analogue de la GnRH en association avec la radiothérapie peuvent avoir plus d’effets secondaires en particulier gastro-intestinaux, liés à la radiothérapie.
Tolérance générale chez la femme (voir rubrique 4.4)
Les effets indésirables les plus fréquents (≥ 10% des patientes) sont la conséquence de la baisse du taux d’œstrogènes. Il s’agit de céphalées, baisse de la libido, troubles du sommeil, changements d’humeur, dyspareunie, dysménorrhée, hémorragie génitale, syndrome d’hyperstimulation ovarienne, hypertrophie ovarienne, douleur pelvienne, douleur abdominale, sécheresse vulvovaginale, hyperhidrose, bouffées de chaleur et asthénie.
Les effets indésirables suivants ont été rapportés et sont considérés comme étant au moins possiblement liés au traitement par la triptoréline. La plupart de ces effets sont connus comme étant liés à une castration biochimique ou chirurgicale. La fréquence de ces effets indésirables peut être classée comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100). Aucune fréquence ne peut être appliquée aux effets indésirables rapportés après la commercialisation. Ils sont donc rapportés avec la mention « fréquence indéterminée ».
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Fréquence indéterminée |
|
Affections du système immunitaire |
|
Hypersensibilité |
|
Choc anaphylactique |
|
Troubles du métabolisme et de la nutrition |
|
|
Appétit diminué Rétention liquidienne |
|
|
Affections psychiatriques |
Troubles du sommeil (incluant insomnie) Trouble de l'humeur Diminution de la libido |
Dépression* Nervosité |
Labilité affective Anxiété Dépression** Désorientation |
Etat confusionnel |
|
Affections du système nerveux |
Céphalée |
Sensation vertigineuse |
Dysgueusie Hypo-esthésie Syncope Atteinte de la mémoire Perturbation de l'attention Paresthésie Tremblement |
Convulsions**** |
|
Affections oculaires |
|
|
Sécheresse oculaire Défauts visuels |
Perturbation visuelle |
|
Affections endocriniennes |
|
|
|
Apoplexie hypophysaire*** |
|
Affections de l'oreille et du labyrinthe |
|
|
Vertige |
|
|
Affections cardiaques |
|
|
Palpitations |
|
|
Affections vasculaires |
Bouffées de chaleur |
|
|
Hypertension |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Dyspnée Epistaxis |
|
|
Affections gastro-intestinales |
|
Nausée Douleur abdominale Gêne abdominale |
Distension abdominale Bouche sèche Flatulence Ulcération buccale Vomissement |
Diarrhée |
|
Affections de la peau et du tissu sous-cutané |
Acné Hyperhidrose Séborrhée |
|
Alopécie Sécheresse cutanée Hirsutisme Onychoclasie Prurit Rash |
Œdème angioneurotique Urticaire |
|
Affections musculo-squelettiques |
|
Arthralgie Spasme musculaire Douleur dans les membres |
Dorsalgie Myalgie |
Faiblesse musculaire |
|
Affections des organes de reproduction et du sein |
Affection mammaire Dyspareunie Saignement génital (incluant saignement vaginal, hémorragie de privation) Syndrome d’hyperstimulation ovarienne Hypertrophie ovarienne Douleur pelvienne Sécheresse vulvo-vaginale |
Douleur mammaire |
Saignement pendant les rapports sexuels Cystocèle Trouble menstruel (incluant dysménorrhée, métrorragie et ménorragie) Kyste de l'ovaire Pertes vaginales |
Aménorrhée |
|
Troubles généraux et anomalies au site d'administration |
Asthénie |
Réaction au site d’injection (incluant douleur, gonflement, érythème et inflammation)
Œdèmes périphériques |
|
Fièvre Malaise |
|
Investigations |
|
Poids augmenté |
Poids abaissé |
Phosphatase alcaline sanguine augmentée Pression artérielle augmentée |
* traitement de longue durée. Cette fréquence est basée sur la fréquence observée dans la classe pour tous les agonistes de la GnRH.
** traitement de courte durée. Cette fréquence est basée sur la fréquence observée dans la classe pour tous les agonistes de la GnRH.
***Rapporté après l’administration initiale chez des patients avec un adénome hypophysaire.
**** Après la mise sur le marché, des convulsions ont été signalées chez des patients recevant des analogues de la GnRH, dont la triptoréline.
Une exacerbation des symptômes de l’endométriose (douleurs pelviennes, dysménorrhée), peut être observée très fréquemment (≥ 10%) lors de l’augmentation initiale et transitoire du taux plasmatique d’œstradiol et disparaît en une à deux semaines. La survenue d’hémorragies génitales incluant des métrorragies et des ménorragies peut être observée dans le mois suivant la première injection.
Dans l’infécondité féminine, l’association avec les gonadotrophines peut entraîner une hyperstimulation ovarienne. Une hypertrophie ovarienne, des douleurs pelviennes et/ou abdominales peuvent être observées.
L’utilisation prolongée des analogues de la GnRH peut induire une perte osseuse, facteur de risque d’une éventuelle ostéoporose.
· Cancer du sein
Les effets indésirables les plus fréquents observés lors du traitement par triptoréline pendant une période allant jusqu'à 5 ans en association avec le tamoxifène ou un inhibiteur de l'aromatase dans les études TEXT et SOFT étaient les bouffées de chaleur, les troubles musculosquelettiques, la fatigue, l'insomnie, l'hyperhidrose, la sécheresse vulvovaginale et la dépression.
Les fréquences des effets indésirables rapportés avec la triptoréline en association avec le tamoxifène (N = 2325) ou l'exémestane (N = 2318) sont rapportés dans le tableau suivant. La fréquence de ces effets indésirables peut être classée comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100), rares (≥ 1/10 000 à < 1/1 000).
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
|
Affections du système immunitaire |
|
Hypersensibilité |
|
|
|
Troubles du métabolisme et de la nutrition |
|
Diabète (intolérance au glucose) Hyperglycémie |
|
|
|
Affections psychiatriques |
Insomnie Diminution de la libido Dépression |
|
|
|
|
Affections du système nerveux |
|
|
Ischémie cérébrale Hémorragie du système nerveux central |
|
|
Affections cardiaques |
|
|
Ischémie myocardique |
Prolongation de l’intervalle QT |
|
Affections vasculaires |
Bouffées de chaleur Hypertension |
Embolie |
|
|
|
Affections gastro-intestinales |
Nausées |
|
|
|
|
Affection de la peau et du tissu sous cutané |
Hyperhidrose |
|
|
|
|
Affections musculo-squelettiques et du tissu conjonctif |
Trouble musculo-squelettique Ostéoporose |
Fracture |
|
|
|
Affections du rein et des voies urinaires |
Incontinence urinaire |
|
|
|
|
Affections des organes de reproduction et du sein |
Dyspareunie Sécheresse vulvovaginale |
|
|
|
|
Troubles généraux et anomalies au site d'administration |
Fatigue |
Réaction au site d’injection |
|
|
Les effets indésirables identifiés ci-dessus doivent être considérés en plus des effets indésirables identifiés chez les hommes et les femmes dans les tableaux ci-dessus afin de décrire de manière complète le profil de tolérance pour l'utilisation dans la suppression de la fonction ovarienne (SFO) en association avec l'exémestane ou le tamoxifène.
L'ostéoporose a été rapportée avec une fréquence plus élevée lors de l'utilisation de la triptoréline en association à l'exémestane que lors de l’association au tamoxifène (39% versus 25%) (voir rubrique 4.4).
Les troubles musculosquelettiques et les fractures ont également été plus fréquemment rapportés lors de l’association à l'exémestane que lors de l’association au tamoxifène (respectivement 89% versus 76% et 6,8% versus 5,2%)
L'hypertension a été rapportée comme un effet indésirable très fréquent lors de l’utilisation de la triptoréline en association avec l'exémestane ou le tamoxifène (respectivement 23% et 22%).
L'hyperglycémie et le diabète ont été rapportés comme des effets indésirables fréquents lors de l’utilisation de la triptoréline en association avec l'exémestane ou le tamoxifène (hyperglycémie : 2,6% et 3,4%, diabète : 2,3% et 2,3% respectivement).
Tolérance générale chez l’enfant (voir rubrique 4.4)
La fréquence de ces effets indésirables peut être classée comme suit : très fréquent (≥1/10) ; fréquent (≥1/100, < 1/10) ; peu fréquent (≥1/1 000, <1/100). Aucune fréquence ne peut être appliquée aux effets indésirables rapportés après la commercialisation. Ils sont donc rapportés avec la mention « fréquence indéterminée ».
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Fréquence indéterminée |
|
Affections du système immunitaire |
|
Hypersensibilité |
|
Choc anaphylactique |
|
Troubles du métabolisme et de la nutrition |
|
|
Obésité |
|
|
Affections psychiatriques |
|
|
Humeur modifiée |
Labilité émotionnelle Dépression Nervosité |
|
Affections du système nerveux |
|
Céphalée |
|
Hypertension intracrânienne idiopathique (pseudotumor cerebri) (voir rubrique 4.4) Convulsions* |
|
Affections oculaires |
|
|
Défauts visuels |
Perturbation visuelle |
|
Affections vasculaires |
|
Bouffées de chaleur |
|
Hypertension |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Epistaxis |
|
|
Affections gastro-intestinales |
|
Douleur abdominale |
Vomissement Constipation Nausées |
|
|
Affections de la peau et du tissu sous-cutané |
|
Acné |
Prurit Rash Urticaire |
Œdème angioneurotique |
|
Affections musculo-squelettiques et systémiques |
|
|
Cervicalgie |
Myalgie |
|
Affections des organes de reproduction et du sein |
Saignement génital (incluant hémorragie vaginale, spotting) Hémorragie de privation Hémorragie utérine Pertes vaginales |
|
Douleur mammaire |
|
|
Troubles généraux et anomalies au site d'administration |
|
Réaction au site d’injection (incluant douleur au site d’injection, érythème au site d’injection, inflammation au site d’injection) |
Malaise |
|
|
Investigations |
|
Poids augmenté |
|
Prolactinémie augmentée Pression artérielle augmentée |
* Après la mise sur le marché, des convulsions ont été signalées chez des patients recevant des analogues de la GnRH, dont la triptoréline.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (Ansm) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En cas de surdosage, celui-ci sera traité de façon symptomatique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
La triptoréline est un décapeptide de synthèse analogue de la GnRH naturelle (hormone de libération des gonadotrophines).
Les études conduites dans l'espèce humaine comme chez l'animal ont montré, qu'après une stimulation initiale, l'administration prolongée de triptoréline entraîne une inhibition de la sécrétion gonadotrope, supprimant par conséquent, les fonctions testiculaire et ovarienne.
A la suite de certaines études animales, un autre mécanisme d'action a été évoqué : effet gonadique direct par diminution de la sensibilité des récepteurs périphériques à la GnRH.
Efficacité et sécurité clinique
Cancer de la prostate
L'administration d'une dose quotidienne de triptoréline peut entraîner une élévation initiale des taux sanguins de LH et de FSH, ce qui a pour conséquence une augmentation initiale des taux de testostérone. La poursuite du traitement entraîne une diminution des taux de LH et de FSH conduisant les stéroïdes à des taux de castration, dans un délai de 2 à 3 semaines, aussi longtemps que le produit est administré.
Le traitement est susceptible d'entraîner une amélioration des signes fonctionnels et objectifs.
Chez les patients avec un cancer de la prostate localement avancé, plusieurs études cliniques, randomisées, à long terme, ont démontré le bénéfice d’une privation androgénique associée à la radiothérapie (RT) en comparaison à la RT seule (RTOG 85-31, RTOG 86-10, EORTC 22863, D’Amico et al., JAMA, 2008).
Une étude clinique randomisée de phase III (EORTC 22961), portant sur 970 patients avec un cancer de la prostate localement avancé (principalement T2c-T4, avec des patients T1c à T2b avec un envahissement ganglionnaire régional), a recherché si une radiothérapie associée à une privation androgénique courte (6 mois, n=483) était non-inférieure à une radiothérapie associée à une privation androgénique longue (3 ans, n=487). L'agoniste de la GnRH était la triptoréline (62,2%) ou d'autres agonistes (37,8%), et l'essai n'était pas stratifié sur le type d'agoniste.
Globalement, la mortalité totale à 5 ans était de 19,0% et 15,2%, respectivement dans les groupes "traitement hormonal court" et "traitement hormonal long", soit un risque relatif de 1,42 (IC unilatéral à 95,71%=1,79 ; IC à 95,71%=[1,09 ; 1,85] ; p=0,65 pour la non infériorité et p=0,0082 pour le test post-hoc de différence entre les groupes de traitement). La mortalité à 5 ans spécifiquement liée à la prostate était 4,78% et 3,2% respectivement dans les groupes "traitement hormonal court" et "traitement hormonal long", soit un risque relatif de 1,71 (IC 95%[1,14 à 2,57] ; p=0,002). La qualité de vie, évaluée avec l’échelle QLQ-C30, n’était pas significativement différente entre les deux groupes (p=0,37).
L'analyse post hoc dans le sous-groupe triptoréline va dans le même sens de l'avantage du traitement long par rapport au traitement court sur la mortalité globale (risque relatif de 1,28 ; IC à 95,71%= [0,89 ; 1,84] ; p=0,38 et p=0,08 respectivement pour les tests post-hoc de non-infériorité et de différence entre les groupes de traitement).
L'indication du cancer de la prostate localisé à haut risque est basée sur des études publiées sur l’association de la radiothérapie et des analogues de la GnRH. Les données cliniques de cinq études publiées ont été analysées (EORTC 22863, RTOG 85-31, RTOG 92-02, RTOG 86-10 et D'Amico et al JAMA, 2008). Elles ont toutes démontré un avantage de l’association des analogues de la GnRH avec la radiothérapie. Dans les études publiées, il n’était pas possible de clairement différentier les populations respectives des indications du cancer de la prostate localement avancé et du cancer de la prostate localisé à haut risque.
Chez les patients atteints d’un cancer de la prostate métastatique résistant à la castration, les études cliniques ont démontré le bénéfice de l’ajout des inhibiteurs de la biosynthèse des androgènes comme l’acétate d’abiratérone ou des inhibiteurs de la voie de signalisation des récepteurs aux androgènes comme l'enzalutamide, au traitement par un analogue de la GnRH, comme la triptoréline.
Puberté précoce centrale
L'inhibition de l'hyperactivité gonadotrope hypophysaire se manifeste, dans les deux sexes, par la suppression de la sécrétion d'œstradiol ou de testostérone, par l'abaissement du pic de LH et par une amélioration du rapport Age statural/Age osseux.
La stimulation gonadique initiale peut être responsable de petites hémorragies génitales nécessitant le recours à un traitement d'acétate de médroxyprogestérone ou de cyprotérone.
Endométriose
L'administration prolongée de triptoréline entraîne une suppression de la sécrétion d'œstradiol et ainsi une mise au repos du tissu endométriosique.
Infécondité féminine
L'administration de triptoréline entraîne une inhibition de la sécrétion gonadotrope (FSH et LH). Ce traitement assure donc la suppression du pic intercurrent de LH endogène et permet une folliculogénèse de meilleure qualité ainsi qu'un recrutement folliculaire augmenté.
Fibromes utérins
Les études ont démontré une diminution régulière et marquée du volume de certains fibromes utérins. Cette diminution est maximale au troisième mois de traitement.
Le traitement par triptoréline induit une aménorrhée après le premier mois de traitement chez la plupart des patientes. Il permet la correction d’une éventuelle anémie résultante de ménorragies et/ou de métrorragies.
Cancer du sein
Des études cliniques réalisées chez des femmes non ménopausées atteintes d'un cancer du sein hormonosensible à un stade précoce ont été réalisées avec la triptoréline afin de supprimer la sécrétion ovarienne d'œstradiol, principale source d'œstrogènes. Sur la base d'études réalisées chez des femmes en bonne santé et des femmes atteintes d'endométriose, l'effet de la triptoréline est atteint 3 à 4 semaines après l'administration.
Deux études de phase 3 (SOFT et TEXT) ont exploré le bénéfice de la suppression de la fonction ovarienne (SFO) sur 5 ans en association avec le tamoxifène (T) ou un inhibiteur de l'aromatase (exémestane - E) chez des femmes non ménopausées atteintes d'un cancer du sein hormonosensible à stade précoce.
La triptoréline était le principal traitement utilisé pour obtenir la SFO (91,0% des sujets randomisés dans l'étude SOFT et 100% dans l'étude TEXT). Les 9% de femmes restantes de l'étude SOFT ont subi une ovariectomie bilatérale ou une irradiation ovarienne bilatérale.
Résultats de l'étude SOFT
L'étude SOFT a été conçue pour évaluer la valeur ajoutée de la SFO associée au tamoxifène en tant que traitement adjuvant chez des femmes non ménopausées atteintes d'un cancer du sein hormonosensible à stade précoce.
Un total de 3 047 femmes ont été incluses dans l’analyse (1 015 femmes dans le groupe T + SFO, 1 018 femmes dans le groupe T seul et 1 014 femmes dans le groupe E + SFO).
Lors d'un suivi médian de 67 mois (5,6 ans), le traitement par T + SFO a réduit de façon non statistiquement significative le risque de maladie (évènement DFS) versus le traitement par T seul (HR = 0,83; IC à 95%, 0,66 à 1,04; p = 0,10). La survie sans maladie (DFS) à 5 ans était de 86,6% (IC à 95%, 84,2% à 88,7%) chez les femmes du groupe T + SFO comparativement à 84,7% (IC à 95%, 82,2% à 86,9%) chez les femmes du groupe T seul.
Cependant, après ajustement des covariables pré-définies dans le modèle multivarié de Cox, les femmes ayant reçu un traitement T + SFO ont un risque significativement réduit de maladie (évènement DFS) par rapport aux femmes traitées par T seul, avec une réduction de 22% (HR = 0,78; IC à 95% 0,62 à 0,98, p = 0,03).
Les femmes ayant reçu un traitement T + SFO avaient un risque réduit non significatif de survenue d'un évènement lié au cancer du sein (évènement BCFI) par rapport aux femmes traitées par T seul (HR = 0,81, IC à 95%, 0,63 à 1,03, p = 0,09). Le pourcentage de patientes sans cancer du sein (BCFI) à 5 ans était de 88,4% (IC à 95%, 86,1% à 90,3%) pour les femmes ayant reçu un traitement T + SFO contre 86,4% (IC à 95%, 84,0% à 88,5%) pour les femmes ayant reçu T seul.
Cependant, après ajustement des covariables pré-définies dans le modèle multivarié de Cox, les femmes traitées par T + SFO avaient un risque significativement réduit de survenue d’un évènement lié au cancer du sein (évènement BCFI) par rapport aux femmes traitées par T seul avec une réduction de 25% (HR = 0,75, IC à 95%, 0,59 à 0,96 ; p = 0,02).
Le bénéfice absolu est plus élevé chez les femmes qui ont reçu une chimiothérapie adjuvante. La survie sans maladie (DFS) à 5 ans pour les femmes ayant reçu une chimiothérapie adjuvante était de 80,7% dans le bras T + SFO et de 77,1% dans le bras T seul (HR = 0,82 ; IC à 95%, 0,64 à 1,07) avec un bénéfice absolu de 3,6% pour le bras T + SFO.
En particulier, le bénéfice de l'ajout de la SFO était observé pour la survie sans maladie (DFS) à 5 ans dans une analyse post-hoc pour le sous-groupe des femmes de moins de 40 ans (HR = 0,74 ; IC à 95%, 0,53 ; 1,03) avec un bénéfice absolu de 4,4 % pour le groupe T + SFO comparé au groupe T seul.
Dans l'étude SOFT, les sujets du bras E + SFO avaient une réduction statistiquement significative du risque de maladie (évènement DFS), par rapport aux sujets du bras T seul (HR = 0,68, IC à 95%, 0,53 à 0,86). Le taux de survie sans maladie (DFS) à 5 ans était de 89,0% (IC à 95%, 86,8% à 90,9%) parmi les sujets du bras E + SFO comparativement à 84,7% (IC à 95%, 82,2% à 86,9%) parmi les sujets du bras T seul.
Les sujets traités par E + SFO avaient une réduction statistiquement significative du risque de survenue d’un évènement lié au cancer du sein (évènement BCFI) par rapport aux sujets traités par T seul (HR = 0,64 ; IC à 95%, 0,49 à 0,83). Le pourcentage de patientes sans cancer du sein (BCFI) à 5 ans était de 90,9% (IC à 95%, 88,9% à 92,6%) parmi les sujets ayant reçu E + SFO, comparativement à 86,4% (IC 95%: 84,0% à 88,5%) parmi les sujets traités par T seul.
Les sujets traités par E + SFO ont une réduction statistiquement significative du risque d'une récidive à distance par rapport aux sujets traités par T seul (HR = 0,71 ; IC à 95%, 0,52 à 0,96). Le pourcentage de patientes sans récidive à distance (DRFI) à 5 ans était de 93,0% (IC à 95%, 91,2% à 94,5%) chez les sujets ayant reçu E + SFO comparativement à 90,7% (IC à 95%, 88,6% à 92,4%).
Le bénéfice absolu est plus élevé chez les femmes qui ont reçu une chimiothérapie adjuvante. Le taux de survie sans maladie (DFS) à 5 ans pour les femmes ayant reçu une chimiothérapie adjuvante était de 83,8% dans le bras E + SFO et de 77,1% dans le bras T seul (HR = 0,70, IC à 95% : 0,53 à 0,92), avec un bénéfice absolu de 6,7% pour le bras E + SFO.
Estimation selon la méthode Kaplan-Meier de la survie sans maladie (DFS) chez les femmes ayant reçu une chimiothérapie préalable
|
|
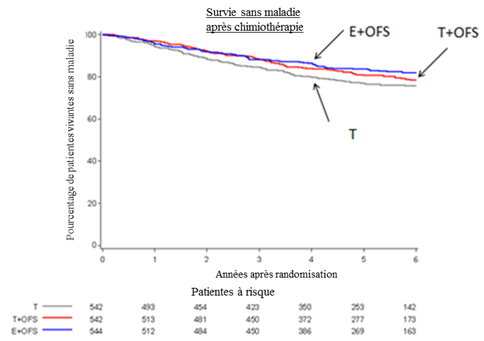
Dans l'étude SOFT à 3 bras, les femmes ayant reçu une chimiothérapie présentaient une proportion plus élevée de critères cliniques associés à un risque élevé de récidive : 49,3% d'âge inférieur à 40 ans, 56,9% avec des ganglions positifs, 47,0% avec une tumeur de taille supérieure à 2 cm et 33,7% avec une tumeur de grade 3.
Résultats combinés des études SOFT et TEXT
L'objectif principal de l'étude TEXT était d'évaluer le rôle des inhibiteurs de l'aromatase (exémestane) chez les femmes traitées par SFO par rapport à aux femmes traitées par SFO+T incluant toutes les femmes des études SOFT et TEXT. Un total de 4690 femmes ont été analysées : 2346 femmes dans le bras E + SFO et 2344 femmes dans le bras T + SFO.
A un suivi médian de 68 mois (5,7 ans), le traitement par E + SFO a permis de réduire de manière statistiquement significative le risque de maladie (évènement DFS) par rapport au traitement par SFO + T (HR = 0,72, IC à 95%, 0,60 à 0,86, p = 0,0002). La survie sans maladie (DFS) à 5 ans était de 91,1% (IC à 95%, 89,7% à 92,3%) pour les femmes traitées par SFO + E contre 87,3% (IC à 95%, 85,7% à 88,7%) pour les femmes traitées par SFO + T.
Estimation selon la méthode Kaplan-Meier de la survie sans maladie (DFS) chez les femmes traitées par SFO+ E versus SFO+T
|
|
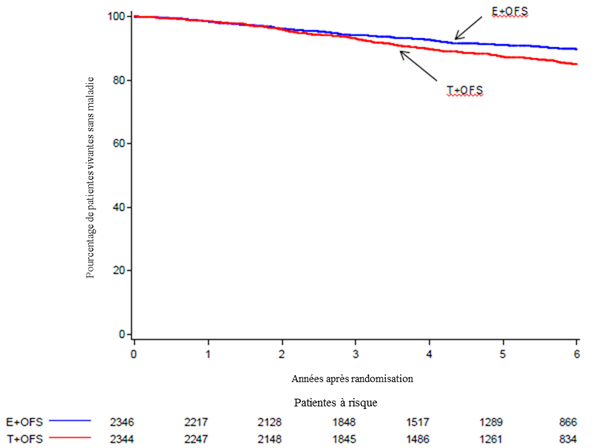
Les femmes traitées par SFO+ E avaient une réduction statistiquement significative du risque de survenue d’un évènement lié au cancer du sein (évènement BCFI) par rapport aux femmes traitées par SFO + T (HR = 0,66; IC à 95%, 0,55 à 0,80; P <0,0001). Le pourcentage de patientes sans cancer du sein (BCFI) à 5 ans à 92,8% (IC à 95%, 91,6% à 93,9%) pour les femmes traitées par SFO + E contre 88,8% (IC à 95%, 87,3% à 90,1%) pour les femmes traitées par SFO + T.
5.2. Propriétés pharmacocinétiques
Après l'injection intramusculaire de DECAPEPTYL L.P. 3 mg chez des femmes atteintes d'endométriose et de fibromes utérins, la concentration sanguine maximale de triptoréline est atteinte entre 2 à 6 heures après l'injection, la valeur du pic est de 11 ng/ml. Il n'y avait aucune preuve d'accumulation du produit après des injections toutes les 4 semaines sur six mois.
Les concentrations plasmatiques minimales sont maintenues entre 0,1 et 0,2 ng/mL. La biodisponibilité du produit à libération prolongée est d'environ 50%.
Ces données observées chez les patientes atteintes d'endométriose et de fibromes utérins peuvent être extrapolées aux patients atteints de cancer du sein car il n’est pas attendu d’impact de la maladie sur les propriétés de libération prolongée du produit.
5.3. Données de sécurité préclinique
La résorption de la poudre est complète en 40 - 45 jours.
La triptoréline n'est pas mutagène in vitro ou in vivo. Aucun effet oncogène n’a été observé chez la souris à des doses de triptoréline allant jusqu’à 6000 microgrammes/kg après 18 mois de traitement. Une étude de carcinogénicité conduite chez le rat pendant 23 mois, a montré une incidence de près de 100% des tumeurs bénignes hypophysaires à chaque dose, conduisant à une mort prématurée. L’augmentation de l’incidence des tumeurs hypophysaires chez le rat est un effet fréquemment associé au traitement par les agonistes de la GnRH. La pertinence clinique de cet effet n'est pas connue.
Composition du solvant : mannitol ; eau pour préparations injectables.
3 ans.
Après reconstitution : à utiliser immédiatement.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Poudre en flacon (verre type I) de 4 ml muni d’un bouchon (élastomère) et d’une capsule (aluminium) + 2 ml de solvant en ampoule (verre) avec seringue et aiguilles. Boîte de 1 flacon et 1 ampoule avec 1 seringue et 2 aiguilles.
6.6. Précautions particulières d’élimination et de manipulation
La suspension pour injection doit être reconstituée en conditions aseptiques et en utilisant exclusivement l’ampoule de solvant pour injection.
Il faut suivre strictement les instructions pour la reconstitution mentionnées ci-après et dans la notice.
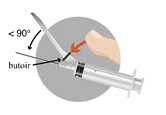
La totalité du solvant doit être aspirée dans la seringue fournie en utilisant l’aiguille pour la reconstitution (20G, sans système de sécurité) et transférée dans le flacon contenant la poudre. La suspension doit être reconstituée en agitant le flacon doucement d’un mouvement circulaire assez longtemps pour obtenir une suspension laiteuse et homogène. Ne pas retourner le flacon.
Il est important de vérifier qu’il n’y a pas d’agglomérats dans le flacon. La suspension obtenue doit être aspirée dans la seringue sans retourner le flacon. L’aiguille pour la reconstitution doit être remplacée par l’aiguille pour injection (20G avec système de sécurité) utilisée pour l’administration du produit.
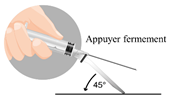
Le produit est sous forme d’une suspension, il doit être injecté dans le muscle fessier immédiatement après la reconstitution pour éviter sa précipitation.
Pour administration unique seulement.
Les aiguilles utilisées, toute suspension non utilisée ou déchet doivent être éliminés conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
70, RUE BALARD
75015 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 339 437 6 9 : Poudre en flacon (verre) + 2 ml de solvant en ampoule (verre) avec seringue et aiguilles ; boîte de 1 flacon et 1 ampoule, 1 seringue et 2 aiguilles.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 11/06/2025
Triptoréline
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours ?
3. Comment utiliser DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours ET DANS QUELS CAS EST-IL UTILISE ?
Ce médicament contient de la triptoréline. La triptoréline appartient à un groupe de médicaments connus sous le nom d’analogues de l’hormone entraînant la libération de gonadotrophines (GnRH). L’une de ses actions est de diminuer la production d’hormones sexuelles dans le corps.
Ce médicament est un analogue d’une hormone naturelle.
Il est utilisé :
Chez l’homme adulte :
· dans le traitement du cancer de la prostate localisé à haut risque ou localement avancé, en association à la radiothérapie.
Chez l’enfant :
· dans le traitement de la puberté qui survient prématurément, c’est-à-dire avant 8 ans chez les filles et 10 ans chez les garçons (puberté précoce centrale).
Chez la femme :
· dans le traitement de l’endométriose.
· dans le traitement de certaines stérilités. Ce médicament est alors généralement associé avec d’autres hormones (appelées gonadotrophines) au cours des procédures de fécondation in vitro (FIVETE).
· dans le traitement pré-opératoire de certains fibromes utérins.
· dans le traitement du cancer du sein hormonosensible au stade précoce chez les femmes non ménopausées ayant reçu une chimiothérapie. DECAPEPTYL L.P. 3 mg est utilisé en association avec des médicaments hormonaux. Vous devrez également prendre :
o un médicament appelé tamoxifène - on vous demandera de prendre ce médicament si vous êtes à haut risque de récidive du cancer
ou
o un médicament «inhibiteur de l'aromatase» tel que l’exémestane. Vous serez traitée avec DECAPEPTYL L.P. 3 mg pendant au moins 6 à 8 semaines avant de commencer à prendre ce médicament.
N'oubliez pas de lire la notice du médicament que vous prenez en association avec DECAPEPTYL L.P. 3 mg.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours ?
· Si vous êtes allergique à la triptoréline, à l’hormone entraînant la libération des gonadotrophines (GnRH), aux autres analogues de la GnRH ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· Si vous êtes enceinte ou si vous allaitez.
· Si vous utilisez DECAPEPTYL L.P. 3 mg pour un cancer du sein, ne prenez pas de médicament appelé « inhibiteur de l’aromatase » (tel que l’exémestane) avant d’avoir déjà été traité pendant au moins 6 à 8 semaines par DECAPEPTYL L.P. 3 mg.
Avertissements et précautions
Adressez-vous à votre médecin pharmacien avant d’utiliser DECAPEPTYL L.P. 3 mg.
· Des dépressions, parfois graves ont été rapportées chez des patients traités par DECAPEPTYL L.P. 3 mg. Si une humeur dépressive apparaît alors que vous êtes traité par DECAPEPTYL L.P. 3 mg, informez votre médecin. Votre médecin peut vouloir surveiller votre dépression pendant le traitement.
· Si vous avez, sans que cela n’ait été découvert jusque-là, une augmentation du volume (tumeur bénigne) de la glande hypophysaire, elle pourrait être découverte au cours du traitement par DECAPEPTYL L.P. 3 mg qui peut entrainer un saignement au niveau de la tumeur (apoplexie hypophysaire). Les symptômes incluent des maux de tête soudains, des vomissements, des problèmes de vue et une paralysie des yeux.
· Si vous prenez des médicaments pour prévenir la coagulation du sang (anticoagulants), des hématomes (bleus) peuvent apparaître au site d’injection.
· Si une convulsion survient, informez immédiatement votre médecin. Des cas de convulsions ont été signalés chez des patients recevant de la triptoréline ou des médicaments similaires. Ces cas sont survenus chez des patients avec ou sans antécédents médicaux d’épilepsie.
Chez l’homme
· Au début du traitement, il y aura une augmentation de la quantité de testostérone dans votre corps. Ceci peut entraîner une aggravation des symptômes du cancer. Contactez votre médecin si cela se produit. Votre médecin pourra vous prescrire des médicaments (un anti-androgène) pour éviter que vos symptômes ne s’aggravent.
· Si vous souffrez d’une obstruction urinaire ou d’une compression de la moelle épinière (nerfs de votre colonne vertébrale) en raison de la propagation de votre cancer de la prostate, votre médecin vous surveillera étroitement pendant les premières semaines du traitement. Si vous ressentez des difficultés à uriner, des douleurs osseuses, une faiblesse des membres inférieurs ou une sensation de picotements, contactez immédiatement votre médecin, qui évaluera votre état de santé et vous traitera de manière appropriée.
· Après castration chirurgicale, la triptoréline n’induit pas de réduction supplémentaire du taux de testostérone.
· Des tests diagnostiques de la fonction gonadotrope hypophysaire ou des organes sexuels conduits durant le traitement et après l’interruption de la thérapie avec DECAPEPTYL L.P. 3 mg peuvent être erronés.
· Chez l’adulte, la triptoréline peut entraîner une perte osseuse (ostéoporose) avec un risque accru de fracture osseuse. Afin de traiter la perte osseuse, votre médecin pourrait vous prescrire un bisphosphonate (médicament utilisé pour traiter la faiblesse osseuse), vous devez donc informer votre médecin si vous présentez l'un des facteurs de risque ci-dessous. Les facteurs de risque peuvent inclure par exemple :
o Si vous ou un membre de votre famille présente une perte osseuse.
o Si vous êtes un grand consommateur d’alcool, et/ou si vous êtes un grand fumeur, ou si vous avez une mauvaise alimentation.
o Si vous suivez un traitement au long cours pouvant provoquer une perte osseuse, comme par exemple des médicaments utilisés pour traiter l’épilepsie ou des stéroïdes (comme l’hydrocortisone ou la prednisolone).
· Vous devez informer votre médecin si vous souffrez de troubles cardiovasculaires, y compris de troubles du rythme cardiaque (arythmie), ou si vous êtes traité par des médicaments pour soigner ces maladies. Le risque de troubles du rythme cardiaque peut être augmenté par l’utilisation de DECAPEPTYL L.P. 3 mg.
· Des contrôles biologiques peuvent être nécessaires au cours du traitement pour vérifier son efficacité.
· Le traitement par des analogues de la GnRH, dont DECAPEPTYL L.P. 3 mg, pourrait augmenter le risque d'anémie (définie comme une diminution du nombre de globules rouges).
Chez la femme
· Chez l’adulte, la triptoréline peut entraîner une perte osseuse (ostéoporose) avec un risque accru de fracture osseuse. Vous devez donc informer votre médecin si vous présentez l'un des facteurs de risque ci-dessous. Les facteurs de risque peuvent inclure par exemple :
o Si vous ou un membre de votre famille présente une perte osseuse.
o Si vous êtes un grand consommateur d’alcool, et/ou si vous êtes un grand fumeur, ou si vous avez une mauvaise alimentation.
o Si vous suivez un traitement au long cours pouvant provoquer une perte osseuse, comme par exemple des médicaments utilisés pour traiter l’épilepsie ou des stéroïdes (comme l’hydrocortisone ou la prednisolone).
· En raison du manque d’expérience clinique chez les femmes de moins de 18 ans, la triptoréline n’est pas recommandée chez les adolescentes et les jeunes femmes car elle pourrait provoquer une perte osseuse.
· Durant le premier mois de traitement, vous pouvez avoir des saignements vaginaux. Ensuite, vos règles doivent normalement s’arrêter. Si des saignements se produisent après le premier mois de traitement, parlez-en à votre médecin.
· Vos règles devraient revenir environ 2 mois après la dernière injection.
· En dehors du traitement de l’infertilité, vous devez utiliser une méthode de contraception autre que la « pilule » pendant toute la période de traitement et jusqu’au retour des règles (voir rubrique grossesse et allaitement).
· Si vous présentez des fibromes sous-muqueux (tumeurs bénignes dans le muscle situé sous la paroi de l'utérus), la triptoréline peut provoquer des saignements lorsque les fibromes se rompent au cours des 6 à 10 premières semaines après le début du traitement. Contactez immédiatement votre médecin si vous présentez des saignements ou douleurs importants ou inhabituels.
· Dans le cadre du traitement de l’infertilité, l’association aux gonadotrophines (hormones stimulant les ovaires) peut entraîner une augmentation de la taille des ovaires ou une hyperstimulation ovarienne qui peuvent se manifester par des douleurs pelviennes et/ou abdominales et des difficultés à respirer. Si cela se produit vous devez consulter immédiatement votre médecin.
· Ce traitement doit être administré sous étroite surveillance médicale avec parfois des contrôles biologiques, cliniques et échographiques stricts et réguliers.
Si vous utilisez DECAPEPTYL L.P. 3 mg en association dans le traitement du cancer du sein :
· Si vous présentez un problème qui affecte vos os, comme l'ostéoporose, informez-en votre médecin. Cela peut affecter la manière dont votre médecin décide de vous traiter. Votre médecin effectuera une mesure de votre densité osseuse avant le début du traitement si vous êtes à risque d'ostéoporose et vous surveillera pendant le traitement.
· Si vous souffrez de diabète ou d'hypertension artérielle, informez-en votre médecin. Votre médecin vérifiera votre glycémie et votre tension artérielle pendant le traitement.
· Si vous souffrez de dépression, informez-en votre médecin. Votre médecin peut vouloir surveiller votre dépression pendant le traitement.
· Si vous interrompez votre traitement par triptoréline, vous devez interrompre en même temps l’inhibiteur de l’aromatase (comme l’exémestane).
Chez l’enfant
· Chez les filles traitées pour une puberté précoce, des saignements vaginaux peuvent apparaître au cours du premier mois de traitement.
· A l’arrêt du traitement, les signes de la puberté réapparaîtront.
· Chez les filles, les règles débuteront environ un an après l'arrêt du traitement.
· Les autres causes de puberté précoce devront être écartées par votre médecin.
· La densité des os diminue pendant le traitement, mais elle se normalise après l’arrêt du traitement.
· Une pathologie de la hanche peut survenir après l’arrêt du traitement (épiphysiolyse de la hanche). Elle se traduit par une raideur de la hanche, une boiterie et/ou une douleur aiguë à l'aine irradiant vers la cuisse. Si cela se produit vous devez consulter votre médecin.
· Si votre enfant souffre d'un mal de tête intense ou récurrent, de problèmes de vue et de bourdonnements ou sifflements d'oreilles, contactez immédiatement un médecin (voir rubrique 4).
Si vous ou votre enfant êtes concerné par un des signes cités ci-dessus, parlez-en à votre médecin.
Autres médicaments et DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris un médicament obtenu sans ordonnance.
Chez l’homme :
DECAPEPTYL L.P. 3 mg peut interagir avec des médicaments utilisés pour traiter les problèmes de rythme cardiaque (par exemple quinidine, procaïnamide, amiodarone et sotalol) ou peut augmenter le risque de troubles du rythme cardiaque quand il est utilisé avec d’autres médicaments (par exemple la méthadone (utilisée pour soulager la douleur et en traitement de substitution des pharmacodépendances), la moxifloxacine (un antibiotique), des antipsychotiques (utilisés pour des troubles mentaux graves).
DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours avec des aliments et boissons
Sans objet.
Ce médicament ne doit pas à être utilisé pendant la grossesse ou l’allaitement.
N’utilisez pas DECAPEPTYL L.P. 3 mg si vous envisagez une grossesse (sauf si DECAPEPTYL L.P. 3 mg est utilisé dans le traitement d’une infertilité).
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou votre pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Vous pouvez ressentir des étourdissements, de la fatigue ou avoir des problèmes de vue tels qu’une vision trouble. Ce sont des effets indésirables possibles du traitement ou de la maladie traitée. Si vous ressentez un de ces effets indésirables, vous ne devez pas conduire ou utiliser des machines.
DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. essentiellement « sans sodium ».
3. COMMENT UTILISER DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours ?
Une injection toutes les 4 semaines.
La durée de traitement sera adaptée selon les cas.
Suivre la prescription médicale.
Cancer de la prostate
La dose habituelle est de 1 flacon de DECAPEPTYL L.P. 3 mg administré par injection intra-musculaire (dans le muscle) une fois par mois (toutes les 4 semaines – 28 jours).
Endométriose et fibromes utérins
Le traitement doit débuter dans les 5 premiers jours du cycle. La dose recommandée est une injection dans le muscle (intramusculaire) d’une ampoule une fois par mois (toutes les 4 semaines – 28 jours).
Infertilité
La dose recommandée est une injection intramusculaire à partir du deuxième jour du cycle (début de la phase folliculaire).
Cancer du sein
La dose recommandée de DECAPEPTYL L.P. 3 mg est une injection dans un muscle, toutes les 4 semaines (28 jours). Le traitement peut durer jusqu'à cinq ans.
DECAPEPTYL L.P. 3 mg est utilisé en association avec un médicament appelé tamoxifène ou un « inhibiteur de l'aromatase », comme l’exémestane. Si vous devez prendre un « inhibiteur de l’aromatase », vous serez traitée avec DECAPEPTYL L.P. 3 mg pendant au moins 6 à 8 semaines avant de commencer à le prendre. Vous recevrez au moins 2 injections de DECAPEPTYL L.P. 3 mg (avec un intervalle de 4 semaines entre les injections) avant de commencer à le prendre.
Utilisation chez les enfants :
La dose est ajustée en fonction du poids du patient.
· Enfants de moins de 20 kg : administrer la moitié de la dose (la moitié du volume de la suspension reconstituée) par injection intramusculaire toutes les 4 semaines (28 jours)
· Enfant dont le poids est compris entre 20 et 30 kg : administrer 2/3 de la dose (soit les 2/3 du volume de la suspension reconstituée) par injection intramusculaire toutes les 4 semaines (28 jours)
· Enfants de plus de 30 kg : 1 dose (toute la suspension reconstituée) par injection intramusculaire toutes les 4 semaines (28 jours)
Mode et voie d’administration
Voie intramusculaire.
La poudre doit être mise en suspension immédiatement avant l’injection en utilisant exclusivement le solvant fourni dans le conditionnement. La suspension obtenue ne doit pas être mélangée à d’autres médicaments.
N.B. : Il est important que la préparation de l’injection soit pratiquée rigoureusement selon les instructions données ci-après. Toute injection défectueuse, conduisant à une perte d’une quantité supérieure à celle qui reste normalement dans le dispositif utilisé pour l’injection doit être signalée.
Si vous avez pris plus de DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours que vous n’auriez dû
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez de prendre DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours
Dans tous les cas, contactez votre médecin.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours
· N'arrêtez pas le traitement avec DECAPEPTYL L.P. 3 mg sans en parler avant à votre médecin. Ceci est particulièrement important si vous utilisez DECAPEPTYL L.P. 3 mg avec un inhibiteur de l'aromatase. En effet, l’arrêt du traitement pourrait entrainer une augmentation des taux d'œstrogènes. Votre médecin surveillera vos taux d'œstrogènes pendant votre traitement avec DECAPEPTYL L.P. 3 mg.
· Si vous arrêtez d'utiliser DECAPEPTYL L.P. 3 mg, vous devez également arrêter le traitement par les inhibiteurs de l'aromatase dans le mois qui suit la dernière injection de DECAPEPTYL L.P. 3 mg.
· Chez la femme, reprise de l’activité ovarienne (possibilité d’ovulation, règles).
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Dans de rares cas, une réaction allergique sévère peut se produire (angiœdème, réaction anaphylactique). Contactez immédiatement un médecin, si vous ressentez des symptômes tels que des difficultés à avaler ou à respirer, des sensations vertigineuses, un gonflement des lèvres, du visage, de la gorge ou de la langue, une éruption cutanée.
Chez l’homme
La plupart des effets indésirables sont attendus, en raison du changement du taux de testostérone dans le corps. Ces effets incluent les bouffées de chaleur, l’impuissance et la baisse de la libido.
Très fréquents (concernent plus de 1 patient sur 10)
· Baisse de la libido.
· Fourmillements dans les jambes.
· Bouffées de chaleur.
· Transpiration excessive.
· Douleur dorsale.
· Impuissance.
· Sensation de faiblesse.
Fréquents (concernent de 1 à 10 patients sur 100)
· Anémie (diminution du nombre de globules rouges).
· Réaction allergique.
· Dépression, changements d’humeur, perte de la libido.
· Sensations vertigineuses, maux de tête.
· Tension artérielle élevée.
· Bouche sèche, nausées.
· Douleurs osseuses et musculaires, douleurs des extrémités des membres.
· Douleur dans la partie inférieure de l’abdomen.
· Réactions au site d’injection (rougeur, inflammation et douleur), œdème (accumulation de liquide dans les tissus).
· Augmentation du poids.
Peu fréquents (concernent de 1 à 10 patients sur 1 000)
· Augmentation du nombre de plaquettes dans le sang.
· Perte d’appétit, diabète, goutte (douleur intense et gonflement des articulations, généralement du gros orteil), excès de lipides dans le sang, augmentation de l’appétit.
· Insomnie, irritabilité.
· Fourmillements ou engourdissement.
· Troubles de la vision.
· Bourdonnements d’oreilles, vertige.
· Palpitations.
· Difficulté à respirer, saignements de nez.
· Douleur abdominale, constipation, diarrhée, vomissements.
· Acné, chute des cheveux, rougeur de la peau, démangeaisons, éruption cutanée, urticaire.
· Douleur articulaire, douleur osseuse, crampe musculaire, faiblesse musculaire, douleur musculaire.
· Réveil nocturne pour uriner, difficulté d’uriner.
· Gonflement des seins, douleur des seins, réduction de la taille des testicules, douleur des testicules.
· Etat de sommeil profond, gonflement des chevilles, pieds et doigts, douleur, frissons, somnolence.
· Modification de certains examens sanguins (incluant une augmentation des marqueurs du foie), pression artérielle augmentée, perte de poids.
Rares (concernent de 1 à 10 patients sur 10 000)
· Inflammation du nez, de la gorge.
· Etat confusionnel, baisse de l’activité, euphorie.
· Troubles de la mémoire.
· Sensation anormale dans les yeux, vision anormale.
· Tension artérielle basse.
· Respiration courte en position couchée.
· Ballonnements, anomalies du goût, flatulence.
· Ampoules cutanées, coloration rouge ou violette de la peau.
· Raideur articulaire, gonflement des articulations, raideur musculo-squelettique, arthrose.
· Douleur thoracique, difficulté à rester debout, symptômes pseudo-grippaux, fièvre.
Fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles)
· Réaction allergique grave pouvant provoquer un gonflement du visage, de la langue et du cou, des sensations de vertiges ou des difficultés à respirer (œdème de Quincke, choc anaphylactique).
· Anxiété.
· En cas de présence d’une tumeur hypophysaire, risque accru de saignement au niveau de la tumeur (apoplexie hypophysaire).
· Modifications de l’électrocardiogramme (prolongation de l’intervalle QT).
· Incontinence urinaire.
· Malaise général.
Comme avec les autres analogues de la GnRH, une augmentation du nombre de globules blancs peut être observée avec DECAPEPTYL L.P. 3 mg.
Les patients traités au long cours par analogue de la GnRH en association à la radiothérapie peuvent avoir plus d’effets secondaires en particulier gastro-intestinaux, liés à la radiothérapie.
Chez la femme
La plupart des effets secondaires sont attendus en raison du changement du niveau d'œstrogènes dans l'organisme.
Effets très fréquents (concernent plus de 1 patient sur 10)
· Difficultés à dormir, changements d'humeur, diminution de la libido.
· Maux de tête.
· Bouffées de chaleur.
· Acné, transpiration excessive, peau grasse.
· Affection des seins.
· Douleur pendant ou après les rapports sexuels, saignements génitaux.
· Syndrome d’hyperstimulation ovarienne (se manifestant par une augmentation de la taille des ovaires et une rétention d'eau), augmentation de la taille des ovaires, douleurs dans le bas ventre, sécheresse vaginale.
· Sensation de faiblesse.
Effets secondaires fréquents (concernent de 1 à 10 patients sur 100)
· Réaction allergique.
· Dépression (en cas de traitement de longue durée), nervosité.
· Sensation vertigineuse.
· Nausées, douleur ou gène abdominales.
· Douleurs articulaires, crampes musculaires, douleur dans les bras et les jambes.
· Douleur des seins.
· Réaction au site d'injection (incluant douleur, gonflement, rougeur et inflammation).
· Œdème des chevilles, pieds et doigts.
· Prise de poids.
Effets secondaires peu fréquents (concernent de 1 à 10 patients sur 1 000)
· Appétit diminué.
· Rétention d’eau.
· Variabilité émotionnelle, anxiété, dépression (en cas de traitement de courte durée), altération de la faculté de se repérer dans le temps et dans l'espace.
· Anomalie du goût.
· Diminution de la sensibilité, perte de connaissance temporaire et brutale, perte de mémoire, manque de concentration, fourmillements ou engourdissement, tremblements.
· Sécheresse des yeux, troubles de la vision.
· Vertige.
· Palpitations.
· Difficulté à respirer, saignements de nez.
· Ballonnements, bouche sèche, flatulence, lésion de la bouche, vomissement.
· Chute de cheveux, peau sèche, excès de poils sur le corps, ongles cassants, démangeaisons, éruption cutanée.
· Douleur dorsale, douleur musculaire.
· Saignements pendant les rapports sexuels, descente de la vessie dans le vagin, saignements en dehors des règles, règles douloureuses, règles abondantes, kyste de l’ovaire qui peut provoquer une douleur, pertes vaginales.
· Perte de poids.
Effets secondaires rapportés après la commercialisation (leur fréquence ne peut pas être estimée sur la base des données disponibles)
· Réaction allergique grave pouvant provoquer un gonflement du visage, de la langue et du cou, des sensations de vertige ou des difficultés à respirer (œdème de Quincke, choc anaphylactique).
· Convulsions.
· Etat confusionnel,
· Perturbation visuelle.
· En cas de présence d’une tumeur hypophysaire, risque accru de saignement au niveau de la tumeur.
· Tension artérielle élevée.
· Diarrhée
· Urticaire.
· Faiblesse musculaire.
· Absence de règles.
· Fièvre, malaise général.
· Modification de certains examens sanguins (incluant une augmentation des marqueurs du foie).
Dans le cadre du traitement de l’endométriose, il est possible d’observer en début de traitement une accentuation des troubles qui l’ont motivé (douleurs abdominales, dysménorrhée) mais qui doivent disparaître en 1 à 2 semaines. Ces phénomènes peuvent survenir même si l’effet du traitement est favorable. Il faut cependant absolument en avertir immédiatement votre médecin traitant.
Effets indésirables lors de l’utilisation pour le cancer du sein en association avec le tamoxifène ou un inhibiteur de l'aromatase
Les effets indésirables suivants ont été observés lorsque DECAPEPTYL L.P. 3 mg a été utilisé pour le cancer du sein en association avec le tamoxifène ou un inhibiteur de l'aromatase :
Effets indésirables très fréquents (concernant plus d'un patient sur 10)
· Difficulté à dormir, baisse de la libido, dépression.
· Bouffées de chaleur, tension artérielle élevée.
· Nausées.
· Transpiration excessive.
· Douleurs articulaires et musculaires, ostéoporose.
· Incontinence urinaire.
· Sécheresse vaginale, douleur pendant ou après les rapports sexuels,
· Sensation de grande fatigue.
Effets indésirables fréquents (concernant de 1 à 10 patients sur 100)
· Réaction allergique.
· Diabète, glycémie élevée (hyperglycémie).
· Caillot dans un vaisseau sanguin.
· Fractures osseuses.
· Douleur, ecchymose, rougeur et gonflement au point d'injection.
Effets indésirables peu fréquents (concernant de 1 à 10 patients de 1 000)
· Saignement cérébral.
· Manque d'apport sanguin vers le cerveau ou le cœur.
Effets secondaires rares (concernant de 1 à 10 patients de 10 000)
· Modifications de l’électrocardiogramme (prolongement de l'intervalle QT).
Chez l’enfant
Effets indésirables très fréquents (concernent plus de 1 patient sur 10)
· Saignements vaginaux et pertes vaginales chez les filles.
Effets indésirables fréquents (concernent de 1 à 10 patients sur 100)
· Réaction allergique.
· Maux de tête.
· Bouffées de chaleur.
· Douleur abdominale.
· Acné.
· Réaction au site d'injection (rougeur, inflammation et/ou douleur).
· Prise de poids.
Effets indésirables peu fréquents (concernent de 1 à 10 patients sur 1 000)
· Obésité.
· Changement d’humeur.
· Troubles de la vision.
· Saignements de nez.
· Vomissement, constipation, nausées.
· Démangeaisons, éruption cutanée, urticaire.
· Douleur du cou.
· Douleur des seins.
· Malaise général.
Effets indésirables rapportés après la commercialisation (leur fréquence ne peut pas être estimée sur la base des données disponibles)
· Réaction allergique grave pouvant provoquer un gonflement du visage, de la langue et du cou, des sensations de vertige ou des difficultés à respirer (œdème de Quincke, choc anaphylactique).
· Convulsions.
· Instabilité émotionnelle, dépression, nervosité.
· Vision anormale.
· Tension artérielle élevée.
· Douleurs musculaires.
· Modification de certains examens sanguins (incluant une augmentation de certaines hormones).
· Hypertension intracrânienne idiopathique (augmentation de la pression intracrânienne autour du cerveau, caractérisée par des maux de tête, une vision double et d'autres symptômes visuels, ainsi que des bourdonnements ou des sifflements d'oreilles).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER DECAPEPTYL L.P. 3 mg, poudre et solvant pour suspension injectable (I.M.) forme à libération prolongée sur 28 jours ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
Après reconstitution, une utilisation immédiate est recommandée.
A conserver à une température ne dépassant pas 25 ºC.
N’utilisez pas DECAPEPTYL L.P. 3 mg, si vous constatez des signes visibles de détérioration au niveau de l’emballage ou des blisters. Retournez le médicament chez votre pharmacien.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Triptoréline................................................................................................................... 3,00 mg*
(sous forme de pamoate de triptoréline)
Pour une unité de prise
*Compte tenu des caractéristiques de la forme pharmaceutique, chaque flacon contient une quantité de pamoate de triptoréline correspondant à 4,3 mg de triptoréline.
· Les autres composants sont :
Composition de la poudre : polymère D,L lactide-coglycolide, mannitol ; carmellose sodique ; Polysorbate 80.
Composition du solvant : mannitol, eau pour préparations injectables.
Ce médicament se présente sous forme de poudre et solvant pour suspension injectable (I.M.) à libération prolongée.
Boîte de 1 flacon de poudre + 1 ampoule de solvant.
Titulaire de l’autorisation de mise sur le marché
70 RUE BALARD
75015 PARIS
FRANCE
Exploitant de l’autorisation de mise sur le marché
70 RUE BALARD
75015 PARIS
FRANCE
PARC D’ACTIVITES DU PLATEAU DE SIGNES
83870 SIGNES
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé (voir rubrique 3) :
INSTRUCTIONS D’UTILISATION
|
1 – PREPARATION DU PATIENT AVANT LA RECONSTITUTION DU MEDICAMENT |
|
|
Préparer le patient en désinfectant le muscle fessier au site d’injection. Cette étape doit être réalisée en premier car la suspension doit être injectée immédiatement après la reconstitution. |
|
|
2 – PREPARATION DE L’INJECTION |
|
|
Deux aiguilles sont fournies dans la boîte. · Aiguille 1 : aiguille longue (38 mm) sans système de sécurité à utiliser pour la reconstitution · Aiguille 2 : aiguille longue (38 mm) avec un système de sécurité à utiliser pour l’injection Aiguille 1 – 38 mm – 20 Gauge Aiguille 2 – 38 mm – 20 Gauge |
|
|
|
|
|
La présence de bulles au-dessus du lyophilisat est un aspect normal du produit. Les étapes suivantes doivent être effectuées les unes après les autres sans interruption. |
|
|
2a · Sortir l'ampoule contenant le solvant. Si une partie de la solution est bloquée dans le haut de l’ampoule, la tapoter afin de la faire descendre dans le corps de l’ampoule. · Visser l’Aiguille 1 (sans système de sécurité) sur la seringue. Ne pas retirer le capuchon de l’aiguille à ce stade. · Casser le haut de l’ampoule en positionnant le point face à soi. · Retirer le capuchon de l’Aiguille 1. Insérer l’aiguille dans l’ampoule et prélever la totalité du solvant dans la seringue. Mettre de côté la seringue contenant le solvant. |
|
|
2b · Sortir le flacon contenant la poudre. Tapoter pour ramener au fond du flacon la poudre éventuellement accumulée en haut du flacon. · Retirer le capuchon en plastique du flacon. · Reprendre la seringue contenant le solvant et enfoncer verticalement l’aiguille au travers du bouchon en élastomère du flacon. Injecter le solvant lentement, de façon à, si possible, rincer toute la partie supérieure du flacon. |
|
|
2c · Remonter l'Aiguille 1 au-dessus du niveau du liquide. · Ne pas retirer l’aiguille du flacon. · Agiter le temps nécessaire (au moins 30 secondes) à l’obtention d’une suspension homogène et laiteuse. · Important : Vérifier l’absence d’agglomérats (en cas d’agglomérats poursuivre l’agitation jusqu’à complète homogénéisation). |
|
|
2d · Quand la suspension est homogène, descendre l’aiguille et, sans retourner le flacon, aspirer toute la suspension. Une petite quantité de suspension restera dans le flacon et devra être éliminée. Un excès de poudre et de solvant est prévu lors de la fabrication pour autoriser cette perte lors de la préparation de la seringue. · Enlever l’Aiguille 1 utilisée pour la reconstitution en la saisissant par l’embout coloré. Visser sur la seringue l’Aiguille 2 avec le système de sécurité. · Faire basculer le manchon de protection de l’aiguille vers le corps de la seringue. Le manchon de protection reste dans la position que vous lui donnez. · Enlever le capuchon de l’aiguille. · Amorcer l’aiguille en vidant l’air contenu dans la seringue et injecter immédiatement. |
|
|
3 – INJECTION INTRAMUSCULAIRE |
|
|
· Pour éviter la sédimentation, injecter immédiatement dans la zone désinfectée le plus rapidement possible (dans la minute suivant la reconstitution). |
|
|
4 – APRES UTILISATION |
|
|
· Activer le système de sécurité d’une seule main o Remarque : garder constamment votre doigt derrière le butoir Il existe deux méthodes pour activer le système de sécurité : § Méthode A : pousser le butoir avec votre doigt ou § Méthode B : pousser la gaine de protection sur une surface plane. o Dans les deux cas, appuyer avec un mouvement ferme rapide jusqu’à l’audition d’un clic. o Vérifier visuellement que l’aiguille est complètement prise dans le manchon de protection. · Les aiguilles utilisées, toute suspension non utilisée ou déchet doivent être éliminés conformément à la réglementation en vigueur.
|
Méthode A
Ou
Méthode B
|