Dernière mise à jour le 01/06/2026
WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable
Indications thérapeutiques
WILFACTIN est fabriqué à partir de plasma humain (la partie liquide du sang) et contient la substance active appelée Facteur von Willebrand humain (VWF).
Le VWF joue un rôle dans la coagulation du sang. L’absence de ce facteur, comme dans la maladie de Willebrand, entraine une coagulation du sang moins rapide qu’elle le devrait, et une tendance accrue aux saignements. La substitution de VWF par WILFACTIN restaurera temporairement les mécanismes de coagulation sanguine.
WILFACTIN est indiqué dans la prévention et le traitement des hémorragies chirurgicales ou autres saignements chez les patients atteints de la maladie de Willebrand, lorsque le traitement seul par la desmopressine (DDAVP) est inefficace ou contre-indiqué.
WILFACTIN peut être utilisé chez tous les groupes d’âge.
WILFACTIN ne doit pas être utilisé dans le traitement de l’Hémophilie A.
Présentations
> 1 flacon(s) en verre de 10 g de poudre - 1 flacon(s) en verre de 10 ml de solvant avec dispositif(s) de transfert
Code CIP : 564 513-8 ou 34009 564 513 8 8
Déclaration de commercialisation : 27/01/2004
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon(s) en verre de 5 g de poudre - 1 flacon(s) en verre de 5 ml de solvant avec dispositif(s) de transfert
Code CIP : 585 101-0 ou 34009 585 101 0 6
Déclaration de commercialisation : 02/02/2015
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon(s) en verre de 20 g de poudre - 1 flacon(s) en verre de 20 ml de solvant avec dispositif(s) de transfert
Code CIP : 585 102-7 ou 34009 585 102 7 4
Déclaration de commercialisation : 02/02/2015
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 01/10/2014 | Inscription (CT) | Le service médical rendu par WILFACTIN 100 UI/ml, poudre et solvant pour solution injectable est important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 01/10/2014 | Inscription (CT) | Ces présentations sont des compléments de gamme qui n’apportent pas d’amélioration du service médical rendu (ASMR V, inexistante) par rapport à la présentation déjà inscrite. |
Autres informations
- Titulaire de l'autorisation : LFB-BIOMEDICAMENTS
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 762 403 5
ANSM - Mis à jour le : 23/05/2025
WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Le produit contient environ 100 UI/mL de facteur von Willebrand humain lorsqu'il est reconstitué avec 5 mL (500 UI), 10 mL (1000 UI) ou 20 mL (2000 UI) d'eau pour préparations injectables.
Avant l'ajout d'albumine, l’activité spécifique de WILFACTIN est supérieure ou égale à 60 UI VWF:RCo/mg de protéines.
L'activité (UI) du FVW est déterminée par la méthode de dosage du cofacteur de la ristocétine (VWF:RCo) par rapport à l'étalon international de facteur von Willebrand concentré (OMS).
Le taux de facteur VIII humain (FVIII) contenu dans WILFACTIN est ≤10 UI/100 UI VWF:RCo.
L’activité (UI) en facteur VIII a été déterminée selon la méthode chromogénique de la Pharmacopée Européenne.
Excipients à effet notoire :
Ce médicament contient du sodium :
· Un flacon de 5 mL (500 UI) contient 0,15 mmol (3,4 mg) de sodium.
· Un flacon de 10 mL (1000 UI) contient 0,3 mmol (6,9 mg) de sodium.
· Un flacon de 20 mL (2000 UI) contient 0,6 mmol (13,8 mg) de sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution injectable.
Poudre : blanche ou jaune pâle, sous forme d’une poudre ou d’un solide friable.
Solvant : clair et incolore.
4.1. Indications thérapeutiques
WILFACTIN est indiqué dans le traitement et la prévention des hémorragies et en situation chirurgicale chez les patients atteints de la maladie de Willebrand (MW) quand le traitement seul par la desmopressine (DDAVP) est inefficace ou contre-indiqué.
WILFACTIN peut être utilisé chez tous les groupes d’âge.
WILFACTIN ne doit pas être utilisé dans le traitement de l'Hémophilie A.
4.2. Posologie et mode d'administration
Le traitement substitutif de la maladie de Willebrand doit être pris en charge ou surveillé par un spécialiste de l'hémostase.
Posologie
Généralement, 1 UI/kg de facteur von Willebrand augmente le taux plasmatique de VWF:RCo d'environ 0,02 UI/mL (2 %).
Des taux de VWF:RCo > 0,6 UI/mL (60 %) et de FVIII:C > 0,4 UI/mL (40 %) doivent être atteints.
L'hémostase est généralement assurée lorsque le facteur VIII coagulant (FVIII:C) atteint un taux de 0,4 UI/mL (40 %). Une injection unique de facteur von Willebrand seul induit une remontée progressive du taux de FVIII:C qui n'atteint son maximum que dans un délai de 6 à 12 heures. Elle ne peut pas corriger immédiatement le taux de FVIII:C. Par conséquent, si le taux initial en FVIII:C du patient se situe au-dessous du seuil critique, dans toutes les situations où une correction rapide de l'hémostase est nécessaire, tels que le traitement d'une hémorragie, un traumatisme sévère ou une intervention chirurgicale en urgence, il est nécessaire de co-administrer un facteur VIII associé au facteur von Willebrand, afin de parvenir à un taux de FVIII:C suffisant pour assurer l'hémostase.
Cependant, si une élévation immédiate du taux de FVIII:C n'est pas nécessaire, notamment en cas d'intervention chirurgicale programmée, ou si le taux de base de FVIII:C est suffisant pour assurer l'hémostase, le médecin peut décider de ne pas recourir à la co-administration de FVIII lors de la première injection de VWF.
· Première injection :
Injection d'une dose de 40 à 80 UI/kg de WILFACTIN pour le traitement des hémorragies ou d’un traumatisme, associée à la quantité nécessaire de facteur VIII, calculé selon le taux plasmatique de base de FVIII:C du patient, afin d’atteindre un taux approprié de FVIII:C, immédiatement avant l'intervention ou le plus tôt possible après la survenue de l'épisode hémorragique ou du traumatisme sévère. En cas de chirurgie, la première injection doit être administrée 1 h avant l'intervention.
Une dose initiale de 80 UI/kg de WILFACTIN peut être indiquée, en particulier chez les patients atteints du type 3 de la maladie de Willebrand, pour lesquels des doses supérieures peuvent être nécessaires, afin de maintenir des taux suffisants.
En cas d'intervention programmée, un délai de 12 - 24 heures est recommandé entre la première injection de WILFACTIN et l'acte chirurgical qui sera précédé d'une seconde injection pré-opératoire 1 heure avant l’intervention. Dans ce cas, il n'est pas nécessaire de co-administrer du facteur VIII, dans la mesure où le FVIII:C endogène a généralement atteint un taux critique de 0,4 UI/ml (40 %) avant l'intervention. Toutefois, ce taux doit être vérifié chez chaque patient.
· Injections suivantes :
Le traitement doit être poursuivi, si nécessaire, par WILFACTIN seul, à la dose de 40 - 80 UI/kg par jour, en 1 ou 2 injections, pendant un à plusieurs jours. La dose et la durée du traitement doivent toujours être adaptées au type de chirurgie, à l'état clinique et biologique du patient (VWF:RCo et FVIII:C) et au type et à la gravité de l'accident hémorragique.
· Prophylaxie à long terme :
WILFACTIN peut être administré en prophylaxie à long terme à une posologie qui est déterminée individuellement pour chaque patient. Des posologies entre 40 et 60 UI/kg de WILFACTIN, administrées deux à trois fois par semaine, permettent de diminuer le nombre d'épisodes hémorragiques.
· Traitement ambulatoire :
Le traitement à domicile peut être initié, notamment en cas de saignements mineurs à modérés ou lors d'une prophylaxie à long terme pour prévenir les saignements, avec l'accord du médecin traitant. Le médecin doit s'assurer qu'une formation appropriée est dispensée et que le traitement est évalué à des intervalles prédéfinis.
Population pédiatrique
Pour chaque indication, la posologie dépend du poids corporel. La dose et la durée du traitement doivent être adaptées à l’état clinique du patient et à ses taux plasmatiques de VWF:RCo et de FVIII:C.
o Chez les enfants âgés de moins de 6 ans, la dose initiale peut être déterminée en fonction de la récupération progressive (RP) du patient ; si les données de RP ne sont pas disponibles, une dose initiale de 60 à 100 UI/kg peut être nécessaire, dans l’objectif d’augmenter les taux de VWF:Rco jusqu’à 100 UI/dL.
o Chez les adolescents et les enfants âgés de plus de 6 ans, la posologie est la même que chez les patients adultes.
· Injections suivantes :
Chez les enfants et les adolescents, les doses suivantes doivent être calculées individuellement en fonction de l’état clinique et des taux de VWF:RCo, et ajustées en fonction de la réponse clinique.
En cas d’intervention chirurgicale programmée :
o Chez les enfants âgés de moins de 6 ans, la première dose sera administrée 12 à 24 heures avant la procédure et l’injection suivante pourra être administrée 30 minutes avant la procédure.
o Chez les adolescents et les enfants âgés de plus de 6 ans, la posologie est la même que chez les patients adultes.
· Prophylaxie :
Chez les enfants et les adolescents, la dose et la fréquence des nouvelles administrations doivent être calculées individuellement en fonction de la récupération progressive et des taux de VWF:RCo, et ajustées en fonction de la réponse clinique.
Mode et voie d'administration
Dissoudre la préparation, comme décrit dans la rubrique 6.6.
WILFACTIN doit être administré par voie intraveineuse, à un débit maximum de 4 mL/minute.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Hypersensibilité
Comme avec toute protéine plasmatique administrée par voie intraveineuse, des réactions d’hypersensibilité sont possibles. Les patients doivent être étroitement surveillés pendant toute la durée de l’injection afin de déceler l'apparition de tout symptôme. Les patients doivent être informés des signes précoces de réactions d’hypersensibilité tels que prurit, urticaire généralisée, oppression thoracique, sifflements respiratoires, hypotension artérielle et réaction anaphylactique.
En cas de survenue de ces symptômes, le traitement doit être interrompu immédiatement. En cas de choc anaphylactique, le traitement médical standard devra être instauré.
Agents transmissibles
Les mesures habituelles de prévention du risque de transmission d'agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection clinique des donneurs, la recherche des marqueurs spécifiques d'infection sur chaque don et sur les mélanges de plasma ainsi que la mise en œuvre dans le procédé de fabrication d'étapes efficaces pour l'inactivation / élimination virale.
Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d'agents infectieux ne peut pas être totalement exclu. Ceci s'applique également aux virus inconnus ou émergents ou autres types d'agents infectieux.
Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humain (VIH), le virus de l’hépatite B (VHB) et le virus de l’hépatite C (VHC).
Les mesures prises peuvent être d'efficacité limitée vis-à-vis des virus non-enveloppés tels que le virus de l’hépatite A et le parvovirus B19. L'infection par le parvovirus B19 peut être sévère chez la femme enceinte (infection du fœtus) et chez les personnes atteintes de déficit immunitaire ou avec une augmentation d’érythropoïèse (ex : anémies hémolytiques).
Une vaccination appropriée (hépatites A et B) des patients recevant régulièrement du facteur von Willebrand dérivé du plasma humain est recommandée.
Il est fortement recommandé, lors de chaque administration de WILFACTIN chez un patient, de noter le nom et le numéro du lot du médicament afin de maintenir un lien entre le patient et le lot du produit.
Accidents thrombo-emboliques
WILFACTIN est un facteur von Willebrand à faible teneur en FVIII. Néanmoins, il y a un risque de complications thrombo-emboliques, particulièrement chez les patients ayant des facteurs de risque clinique ou biologique connus. Dans ce cas, ces patients à risque doivent être surveillés par la recherche des signes précoces de thrombose. Une prévention des complications thrombo-emboliques veineuses doit être instaurée, selon les recommandations en vigueur.
Lorsque WILFACTIN est prescrit, le médecin traitant doit être averti que la poursuite du traitement peut entraîner une augmentation excessive du FVIII:C. Par conséquent, chez les patients nécessitant des administrations fréquentes de WILFACTIN, particulièrement en cas d’association avec du facteur VIII, le taux plasmatique de FVIII:C doit être surveillé pour éviter son élévation excessive prolongée, ce qui peut potentialiser le risque de complications thrombo-emboliques.
Immunogénicité
Les patients atteints de la maladie de Willebrand, en particulier ceux de type 3, peuvent développer des anticorps neutralisants (inhibiteurs) du facteur von Willebrand. Si le taux plasmatique attendu de VWF:RCo n'est pas atteint ou si l'hémorragie n'est pas contrôlée à une dose appropriée, des analyses biologiques appropriées doivent être pratiquées afin de rechercher la présence d'un inhibiteur du facteur von Willebrand. Chez les patients présentant des taux élevés d'inhibiteur, le traitement par facteur von Willebrand peut ne pas être efficace et d'autres options thérapeutiques doivent être envisagées.
Considérations relatives aux excipients (teneur en sodium)
Ce médicament contient du sodium.
Si plus de 3300 UI sont injectées (plus de 1 mmol de sodium), cela doit être pris en compte chez les patients suivant un régime hyposodé strict (voir rubrique 2 pour la quantité par flacon).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction médicamenteuse avec le facteur von Willebrand humain n'est connue à ce jour.
4.6. Fertilité, grossesse et allaitement
L'innocuité de WILFACTIN au cours de la grossesse et de l'allaitement n’a pas été évaluée par des essais cliniques.
WilFACTIN ne doit être administré chez les femmes enceintes ou allaitantes, présentant un déficit en facteur von Willebrand, que si clairement indiqué.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Aucun effet sur l'aptitude à conduire des véhicules ou à utiliser des machines n’a été observé.
Les effets indésirables suivants peuvent survenir au cours du traitement par WILFACTIN :
Réactions allergiques et anaphylactiques (y compris l’état de choc dans de rares cas), événements thromboemboliques (principalement chez les patients présentant des facteurs de risque), formation d'inhibiteurs contre le VWF et réactions au site d'administration.
Liste tabulée des effets indésirables
Le tableau ci-dessous présente un résumé des effets indésirables observés dans 6 essais cliniques et une étude post-commercialisation non interventionnelle, ainsi que provenant d'autres sources post-commercialisation. Au cours des études, 226 patients ont été exposés à WILFACTIN pour un total de 16 640 jours d'exposition.
Les effets indésirables ont été classés selon la classification MedDRA (classes de systèmes d'organes), les termes préférentiels et la fréquence.
Les fréquences de survenue des effets indésirables ont été estimées comme suit : très fréquent (³1/10) ; fréquent (³1/100 à <1/10) ; peu fréquent (³1/1000 à <1/100) ; rare (³1/10 000 à <1000) ; très rare (<1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Pour les effets indésirables rapportés spontanément après commercialisation, la fréquence de notification est classée comme indéterminée.
|
Classe de systèmes d’organes MedDRA |
Effet indésirable (Terme préférentiel) |
Fréquence par nombre de patients |
|
Affections hématologiques et du système lymphatique |
Inhibition du facteur de von Willebrand* |
Fréquence indéterminée |
|
Affections du système immunitaire |
Hypersensibilité |
Peu fréquent |
|
Choc anaphylactique* |
Fréquence indéterminée |
|
|
Affections du système nerveux |
Sensation vertigineuse |
Peu fréquent |
|
Paresthésie, Hypoesthésie |
Peu fréquent |
|
|
Affections vasculaires |
Bouffée de chaleur |
Peu fréquent |
|
Événements thromboemboliques* |
Fréquence indéterminée |
|
|
Affections de la peau et du tissu sous-cutané |
Prurit |
Peu fréquent |
|
Troubles généraux et anomalies au site d’administration |
Réactions au site d'administration** (y compris réaction au site de perfusion, inflammation au site de perfusion et inflammation au site de ponction vasculaire) |
Fréquent |
|
Sensation d’oppression |
Peu fréquent |
|
|
Frissons, sensation de froid |
Peu fréquent |
|
|
Fièvre* |
Fréquence indéterminée |
* Signalé lors de la surveillance post-commercialisation à une fréquence « indéterminée », par convention.
** Termes de haut niveau (High Level Group) du dictionnaire MedDRA.
Description de certains effets indésirables
Des réactions d’hypersensibilité ou allergiques (telles que angioedème, brûlures ou picotements au point d'injection, frissons, bouffées congestives, urticaire généralisée, céphalées, prurit, hypotension artérielle, lipothymie/malaise, léthargie, nausées, agitation, tachycardie, oppression thoracique, fourmillements, vomissements, sifflements respiratoires), ont été observés peu fréquemment et peuvent, dans certains cas, évoluer vers une réaction anaphylactique sévère (voire état de choc).
L'apparition d'un anticorps neutralisant (inhibiteur) du facteur von Willebrand, en particulier chez les patients atteints du type 3 de la maladie, est très rare. Si de tels inhibiteurs se développent, leur présence se manifeste par une réponse clinique insuffisante. La présence d’anticorps anti-facteur von Willebrand peut être étroitement corrélée à des réactions anaphylactiques. Par conséquent, la recherche d'inhibiteurs doit être effectuée chez tout patient présentant une réaction anaphylactique. Dans de tels cas, il est recommandé de contacter un centre spécialisé dans l'hémophilie.
WILFACTIN est un facteur von Willebrand à faible teneur en FVIII. Néanmoins, il y a un risque de complications thrombo-emboliques, particulièrement chez les patients ayant des facteurs de risque clinique ou biologique connus. Par conséquent, les patients à risque doivent être surveillés.
Pour des informations sur la sécurité vis-à-vis des agents transmissibles, voir rubrique 4.4.
WILFACTIN a été évalué chez 56 patients âgés de moins de 18 ans, dont 23 âgés de moins de 6 ans, 21 âgés de 6 à 11 ans et 12 âgés de plus de 11 ans.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Aucun symptôme lié à un surdosage avec le facteur von Willebrand n'a été rapporté.
Des évènements thrombo-emboliques peuvent apparaître en cas de surdosage important.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
WILFACTIN agit comme le facteur von Willebrand endogène.
L'apport de facteur von Willebrand permet de corriger les troubles de l'hémostase observés chez les malades atteints d’un déficit en facteur von Willebrand (maladie de Willebrand). Son effet est double :
· il rétablit l'adhésion des plaquettes au sous-endothélium vasculaire au niveau de la lésion (le facteur von Willebrand ayant la propriété de se lier à la fois au sous-endothélium vasculaire et à la membrane plaquettaire) ce qui assure l'hémostase dite primaire, comme en témoigne le raccourcissement fréquemment observé du temps de saignement. Cet effet apparaît immédiatement et est connu pour être en grande partie fonction du degré de multimérisation du principe actif ;
· il a une action retardée dans les déficits associés en facteur VIII. Après administration intraveineuse, il se fixe au facteur VIII endogène (produit normalement par le patient) et le stabilise en évitant que ce dernier ne soit rapidement dégradé. Par conséquent, l’administration de facteur von Willebrand pur (préparation de VWF à faible taux de FVIII) normalise le taux de FVIII:C comme effet résultant de la première injection. Cet effet est durable et persiste lors des injections ultérieures. L’administration de préparations avec de VWF contenant du FVIII :C restaure le taux de FVIII:C immédiatement après la première injection.
5.2. Propriétés pharmacocinétiques
Une étude de pharmacocinétique avec WilFACTIN réalisée chez 8 patients adultes atteints de la maladie de Willebrand de type 3 a montré que pour FVW:RCo :
· L'AUC0-∞ moyenne est de 3444 UI.h/dL après une dose unique de 100 UI/kg de WILFACTIN,
· La récupération moyenne est de 2,1 [UI/dL]/[UI/kg] de la préparation injectée,
· La demi-vie se situe entre 8 et 14 heures, avec une valeur moyenne de 12 heures,
· La clairance moyenne est de 3,0 mL/h/kg.
Le pic plasmatique du facteur von Willebrand est généralement atteint entre 30 minutes et 1 heure après l'injection.
La normalisation du taux de FVIII est progressive et nécessite normalement un délai variable de 6 à 12 heures. Cet effet est prolongé durant 2 à 3 jours.
L'augmentation du taux de FVIII est progressive et la normalisation nécessite un délai variable de 6 à 12 heures. L'élévation du taux de FVIII est en moyenne de 6 % (UI/dL) par heure. Ainsi, même chez ces patients dont le taux initial de FVIII:C est inférieur à 5 % (UI/dL), un taux de FVIII:C d'environ 40 % (UI/dL) est atteint dès la 6e heure après injection et se maintient durant 24 heures.
Données pédiatriques
Le profil pharmacocinétique après injection de WILFACTIN n’a pas fait l’objet d’une description complète (Cmax, Tmax, AUC, clairance, demi-vie et récupération moyenne) chez la population pédiatrique âgée de moins de 18 ans.
Chez 7 enfants âgés de moins de 6 ans (2 âgés de 28 jours à 23 mois et 5 âgés de 24 mois à 6 ans) présentant une maladie de Willebrand sévère (5 de type 3, 1 de type 1 et 1 de type 2), après une perfusion moyenne de 101,1 ± 5,0 UI/kg, une récupération progressive moyenne du VWF:RCo de 1,75 ± 0,35 (UI/dL)/(UI/kg) a été observée 15 minutes après la perfusion, avec une variabilité inter-individuelle importante (plage de 1,14 à 2,03). Les analyses de contrôle de la récupération initiale et à 6 mois, après exposition à un traitement d’une durée de 3 à 9 jours, étaient évaluables chez seulement quatre de ces enfants. Le taux moyen de la récupération était de 0,87 ± 0,12 (UI/dL)/(UI/kg), (plage de 0,7 à 1,0).
5.3. Données de sécurité préclinique
Les études de toxicité par administration réitérée ne sont pas réalisables, en raison de l’induction d’anticorps dirigés contre les protéines hétérologues des modèles animaux.
Les données précliniques ne laissent supposer aucune potentialité mutagène de WILFACTIN.
albumine humaine,
chlorhydrate d'arginine,
glycine,
citrate de sodium et
chlorure de calcium.
Solvant : eau pour préparations injectables.
Seuls les dispositifs d'injection en polypropylène autorisés doivent être utilisés, car l'adsorption du Facteur von Willebrand humain sur les surfaces internes de certains matériels d’injection peut être responsable de l'échec du traitement.
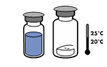
La stabilité physico-chimique en cours d’utilisation a été démontrée pendant 24 heures à 25 °C.
D’un point de vue microbiologique, il est recommandé d’utiliser le produit immédiatement.
6.4. Précautions particulières de conservation
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Poudre en flacon (verre de type I) muni d'un bouchon (bromobutyle) et d'une capsule de protection + 5 mL de solvant en flacon (verre de type I ou de type II) muni d'un bouchon (bromobutyle ou chlorobutyle) et d'une capsule de protection avec un système de transfert - boîte de 1.
Poudre en flacon (verre de type I) muni d'un bouchon (bromobutyle) et d'une capsule de protection + 10 mL de solvant en flacon (verre de type I ou de type II) muni d'un bouchon (bromobutyle ou chlorobutyle) et d'une capsule de protection avec un système de transfert - boîte de 1.
Poudre en flacon (verre de type I) muni d'un bouchon (bromobutyle) et d'une capsule de protection + 20 mL de solvant en flacon (verre de type I ou de type II) muni d'un bouchon (bromobutyle ou chlorobutyle) et d'une capsule de protection avec un système de transfert - boîte de 1.
6.6. Précautions particulières d’élimination et de manipulation
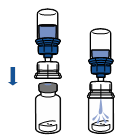
Reconstitution :
Les règles d'asepsie en vigueur doivent être appliquées. Le système de transfert n'est utilisé que pour reconstituer le médicament, comme décrit ci-dessous. Il n'est pas destiné à l'administration du médicament au patient.
|
|
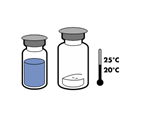
· Amener les deux flacons (poudre et solvant) à une température ne dépassant pas 25 °C.
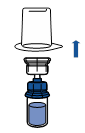
· Retirer la capsule protectrice du flacon de solvant (eau pour préparations injectables) et du flacon de poudre.
· Désinfecter la surface de chaque bouchon.
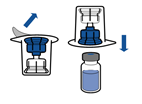
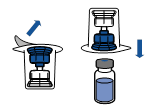
· Retirer l’opercule du dispositif Mix2Vial. Sans extraire le dispositif de son emballage, enclencher l’extrémité bleue du Mix2Vial sur le bouchon du flacon de solvant.
· Retirer puis jeter l’emballage. Prendre soin de ne pas toucher la partie maintenant exposée du dispositif.
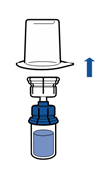
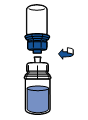
· Retourner l’ensemble flacon de solvant-dispositif et l’enclencher sur le flacon de poudre par la partie transparente du dispositif. Le solvant est transféré automatiquement dans le flacon de poudre. Maintenir l’ensemble et agiter doucement, d’un mouvement circulaire, pour dissoudre totalement le produit.
· En maintenant la partie produit reconstituée d’une main et la partie solvant de l’autre, séparer les flacons en dévissant le dispositif Mix2Vial.
|

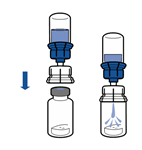

La poudre est généralement dissoute instantanément et devrait dissoudre en moins de 5 minutes.
Administration :
|
|
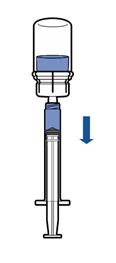
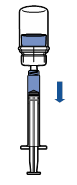
· Tenir le flacon de produit reconstitué verticalement, en vissant une seringue stérile sur le dispositif Mix2Vial. Aspirer lentement le produit dans la seringue. · Une fois le produit transféré dans la seringue, tenir celle-ci fermement (piston dirigé vers le bas), dévisser le dispositif Mix2Vial et le remplacer par une aiguille intraveineuse ou une aiguille épicrânienne. · Expulser l’air de la seringue et piquer la veine après désinfection. · Injecter lentement par voie intraveineuse en une seule fois, immédiatement après reconstitution, sans dépasser un débit maximum de 4 mL/minute
|
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Le produit reconstitué doit être inspecté visuellement pour vérifier la présence éventuelle de particules ou d’une décoloration avant administration. La solution obtenue est limpide ou légèrement opalescente, incolore ou faiblement jaunâtre.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
3, AVENUE DES TROPIQUES
ZA DE COURTABŒUF
91940 LES ULIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 585 101-0 ou 34009 585 101 0 6 : 5 g de poudre en flacon (verre) + 5 mL de solvant en flacon (verre) + un système de transfert-boîte de 1.
· 564 513-8 ou 34009 564 513 8 8 : 10 g de poudre en flacon (verre) + 10 mL de solvant en flacon (verre) + un système de transfert - boîte de 1.
· 585 102-7 ou 34009 585 102 7 4 : 20 g de poudre en flacon (verre) + 20 mL de solvant en flacon (verre) + un système de transfert-boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 23/05/2025
WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable
Facteur von Willebrand humain
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable et dans quel cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable ?
3. Comment utiliser WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable ?
6. Contenu de l’emballage et autres informations
1. QU’EST-CE QUE WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
WILFACTIN est fabriqué à partir de plasma humain (la partie liquide du sang) et contient la substance active appelée Facteur von Willebrand humain (VWF).
Le VWF joue un rôle dans la coagulation du sang. L’absence de ce facteur, comme dans la maladie de Willebrand, entraine une coagulation du sang moins rapide qu’elle le devrait, et une tendance accrue aux saignements. La substitution de VWF par WILFACTIN restaurera temporairement les mécanismes de coagulation sanguine.
WILFACTIN est indiqué dans la prévention et le traitement des hémorragies chirurgicales ou autres saignements chez les patients atteints de la maladie de Willebrand, lorsque le traitement seul par la desmopressine (DDAVP) est inefficace ou contre-indiqué.
WILFACTIN peut être utilisé chez tous les groupes d’âge.
WILFACTIN ne doit pas être utilisé dans le traitement de l’Hémophilie A.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable ?
· Si vous êtes allergique au Facteur von Willebrand humain ou à l'un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
· Si vous souffrez d’Hémophilie A.
Avertissements et précautions
Le suivi de votre traitement par WILFACTIN doit toujours être effectué par un médecin spécialiste des troubles de l’hémostase.
· Si vous avez des saignements importants et qu’un examen sanguin montre que votre taux sanguin de facteur VIII est réduit, vous recevrez en plus du VWF, une préparation de facteur VIII dans les douze premières heures.
Réactions allergiques
Comme avec tout médicament contenant des protéines dérivées du sang ou du plasma et injecté dans une veine (administration intraveineuse), des réactions allergiques (hypersensibilité) peuvent survenir.
Pendant l'injection, soyez attentif à tout signe précoce d'hypersensibilité. Ceux-ci comprennent des éruptions cutanées (urticaire ou urticaire généralisée), une oppression thoracique, une respiration sifflante, une chute de la pression sanguine (hypotension) et des réactions allergiques graves (anaphylaxie).
Votre médecin vous informera des symptômes annonciateurs d’une réaction allergique.
En cas d’apparition de signes ou de symptômes d’hypersensibilité, le traitement doit être interrompu et vous devez consultez un médecin immédiatement.
Sécurité virale
Lorsque des médicaments sont préparés à partir de sang ou de plasma humain, certaines mesures sont mises en place afin de prévenir les infections pouvant être transmises aux patients. Celles-ci comprennent :
· Une sélection rigoureuse des donneurs de sang et de plasma de façon à exclure les donneurs risquant d’être porteurs d’infections,
· Le contrôle de chaque don et des mélanges de plasma pour détecter la présence de virus/d’infections,
· L’inclusion dans le procédé de fabrication d’étapes capables d’inactiver ou d'éliminer les virus.
Malgré ces mesures, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission de maladies infectieuses ne peut être totalement exclu. Ceci s’applique également à tous les virus inconnus ou émergents ou autres types d’agents infectieux.
Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés comme le virus de l'immunodéficience humaine (VIH ou virus du SIDA), le virus de l'hépatite B et le virus de l'hépatite C.
Ces mesures peuvent être d'efficacité limitée vis-à-vis des virus non enveloppés tels que le virus de l'hépatite A et le parvovirus B19. L'infection par le parvovirus B19 peut être sévère chez la femme enceinte (car il existe un risque d'infection de l'enfant à naître) et chez les personnes atteintes d’un déficit immunitaire ou de certains types d'anémie (par exemple, l’anémie falciforme ou l'anémie hémolytique).
Vaccinations
Votre médecin peut vous recommander une vaccination appropriée contre les hépatites A et B si vous recevez de manière régulière/répétée du facteur von Willebrand dérivé de plasma humain.
Enregistrement du numéro de lot
Il est fortement recommandé, lors de chaque administration de WILFACTIN, d'enregistrer le nom et le numéro du lot du médicament afin d’assurer la traçabilité de chaque lot utilisé.
Risque thrombotique
Les vaisseaux sanguins peuvent également être obstrués par des caillots sanguins (thromboses).
Ce risque existe particulièrement si vos antécédents médicaux ou résultats biologiques indiquent que vous présentez certains facteurs de risque.
Dans ce cas, vous serez surveillé très attentivement pour détecter les premiers signes de thrombose, et un traitement préventif (prophylaxie), contre l’obstruction des veines par des caillots sanguins, devra être instauré.
Lors de l’utilisation d’un facteur von Willebrand contenant du facteur VIII, votre médecin doit être conscient que la poursuite du traitement peut entraîner une augmentation excessive en FVIII. Si vous recevez un tel VWF contenant du FVIII, votre médecin doit surveiller régulièrement votre taux plasmatique de FVIII. Cela garantit que votre taux plasmatique de FVIII ne reste pas à un niveau excessif, ce qui pourrait augmenter le risque d'événements thrombotiques.
Efficacité limitée
Des protéines neutralisant l'effet du VWF peuvent se former chez les patients atteints de la maladie de Willebrand, et en particulier chez les patients de type 3. Ces protéines sont appelées anticorps neutralisants ou inhibiteurs. Si les résultats biologiques montrent que votre taux de VWF ne se normalise pas, ou si le saignement ne s'arrête pas malgré une dose suffisante de WILFACTIN, votre médecin vérifiera si des inhibiteurs du VWF se forment dans votre corps. Si de tels inhibiteurs sont présents en concentration élevée, le traitement par VWF peut ne pas être efficace et d'autres options thérapeutiques doivent être envisagées. Le nouveau traitement sera prescrit par un médecin spécialisé dans le traitement des troubles de l’hémostase.
Une vaccination appropriée (hépatites A et B) des patients recevant des facteurs de coagulation est recommandée.
Autres médicaments et WILFACTIN
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Grossesse et allaitement
WILFACTIN ne doit être utilisé pendant la grossesse et l'allaitement que si cela est clairement indiqué.
L’innocuité de WILFACTIN au cours de la grossesse et de l’allaitement n’a pas été étudiée au cours d’études cliniques. Les études chez l’animal sont insuffisantes pour établir la sécurité vis-à-vis de la fertilité, la grossesse et le développement de l’enfant pendant la grossesse et après la naissance.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Aucun effet sur l'aptitude à conduire des véhicules ou à utiliser des machines n’a été observé.
WILFACTIN contient du sodium
Un flacon de 5 mL (500 UI) de WILFACTIN contient 0,15 mmol (3,4 mg) de sodium.
Cela correspond à 0,17 % de l'apport journalier maximal conseillé en sodium pour un adulte.
Un flacon de 10 mL (1000 UI) de WILFACTIN contient 0,3 mmol (6,9 mg) de sodium.
Cela correspond à 0,35 % de l'apport journalier maximal conseillé en sodium pour un adulte.
Un flacon de 20 mL (2000 UI) de WILFACTIN contient 0,6 mmol (13,8 mg) de sodium.
Cela correspond à 0,69 % de l'apport journalier maximal conseillé en sodium pour un adulte.
3. COMMENT UTILISER WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable ?
Si votre médecin pense que l'administration pourrait être effectuée à votre domicile, il/elle vous fournira des instructions appropriées.
Dose
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin. Vérifiez auprès de votre médecin en cas de doute.
De préférence, WILFACTIN doit être administré par votre médecin ou votre infirmier/ère. Cependant, si WILFACTIN vous a été prescrit pour une utilisation à domicile, votre médecin veillera à ce qu'on vous montre comment l'injecter et quelle quantité utiliser. Veillez à suivre les instructions données par votre médecin et demandez de l'aide si vous avez des problèmes pour manipuler la seringue. Toute personne utilisant la seringue doit avoir reçu une formation appropriée.
Votre médecin calculera votre dose de WILFACTIN à utiliser (en unités internationales ou UI).
La dose dépend :
· du poids corporel,
· du site de saignement,
· de l’intensité du saignement,
· de votre condition clinique,
· de la chirurgie que vous allez subir,
· du niveau d’activité du VWF dans votre sang après la chirurgie,
· de la sévérité de votre maladie.
La dose varie de 40 à 80 UI/kg.
Au cours du traitement, votre médecin vous recommandera des tests sanguins pour contrôler :
· le taux de facteur VIII (FVIII:C),
· le taux de facteur von Willebrand (VWF:RCo),
· la présence d’un inhibiteur,
· les premiers signes de formation de caillots si vous avez un risque de développer ces complications.
Selon les résultats de ces tests, votre médecin pourra décider d'adapter la dose et la fréquence de vos injections.
Dans certains cas, l’utilisation de préparation de facteur VIII (une autre protéine de la coagulation) en plus de WILFACTIN est nécessaire pour traiter ou prévenir plus rapidement les saignements (lors de situations d’urgences ou de saignements aigus).
WILFACTIN peut également être administré en prophylaxie à long terme, dans ce cas, la dose sera également déterminée individuellement. Des doses allant de 40 à 60 UI/kg de WILFACTIN, administrées deux ou trois fois par semaine, permettent de diminuer le nombre d'épisodes hémorragiques.
Utilisation chez les enfants et les adolescents
Chez les enfants et les adolescents, la posologie dépend du poids corporel. Dans certains cas, notamment chez les patients les plus jeunes (âgés de moins de 6 ans), des doses plus élevées (allant jusqu’à 100 UI/kg) peuvent être nécessaires.
Si vous avez l’impression que l’effet de WILFACTIN est trop fort ou trop faible, consultez votre médecin.
Méthode d'administration
Des instructions détaillées sur la reconstitution et l’administration du médicament sont données à la fin de la notice.
Si vous avez utilisé plus de WILFACTIN, poudre et solvant pour solution injectable, que vous n’auriez dû
Aucun symptôme de surdosage n’a été rapporté avec WILFACTIN.
Cependant, le risque de thrombose ne peut être exclu en cas de surdosage important.
Si vous oubliez d’utiliser WILFACTIN, poudre et solvant pour solution injectable
Si vous oubliez d'utiliser WILFACTIN, demandez conseil à votre médecin.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, votre pharmacien ou votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Veuillez contacter immédiatement votre médecin si :
· Vous remarquez des symptômes d’hypersensibilité ou de réactions allergiques (observés peu fréquemment : peuvent affecter jusqu’à 1 personne sur 100).
· Dans certains cas, ces réactions peuvent évoluer vers une réaction allergique sévère (anaphylaxie) voire un choc anaphylactique (observé avec une fréquence inconnue).
Les signes d'alerte d'une réaction allergique sont :
o Difficultés à respirer et à avaler
o Respiration sifflante
o Oppression thoracique
o Augmentation de la fréquence cardiaque
o Diminution ou chute de la pression sanguine
o Evanouissement
o Fatigue extrême
o Agitation, nervosité
o Maux de tête
o Frissons, sensation de froid
o Bouffée congestive, bouffées de chaleur
o Gonflement de différentes parties du corps
o Eruption cutanée, urticaire généralisée
o Brûlures et picotements au point d'injection
o Fourmillements
o Vomissements
o Nausées
· Vous remarquez que le médicament cesse de fonctionner correctement (le saignement n'est pas contrôlé). Cela peut être dû à l'inhibition du facteur von Willebrand (observée avec une fréquence inconnue).
Des protéines qui neutralisent l'effet du VWF peuvent se former chez les patients atteints de la maladie de Willebrand, et en particulier chez les patients de type 3. Ces protéines sont appelées anticorps neutralisants ou inhibiteurs. Les patients traités par VWF doivent être attentivement surveillés par leurs médecins sur le plan clinique et biologique en ce qui concerne le développement de ces inhibiteurs. Si de tels inhibiteurs se développent, leur présence peut se manifester par une réponse clinique insuffisante ou survenir en même temps que des réactions allergiques graves.
· Vous remarquez des symptômes liés à une irrigation altérée de vos extrémités (par exemple, des extrémités froides et pâles) ou des organes vitaux (par exemple, une douleur thoracique sévère). Cela peut être dû à la formation de caillots sanguins dans les vaisseaux (observés avec une fréquence inconnue).
Il existe un risque de formation de caillots sanguins (thrombose), en particulier chez les patients présentant des facteurs de risque connus. Après correction du déficit en facteur von Willebrand, vous devez être surveillé pour des signes précoces de thrombose ou de coagulation intravasculaire disséminée et recevoir un traitement pour prévenir la thrombose dans les situations impliquant un risque accru de thrombose (après une chirurgie, pendant un alitement, en cas de déficit en inhibiteur de la coagulation ou en enzyme fibrinolytique).
Si vous recevez des préparations de VWF contenant du FVIII, le risque de thrombose peut également augmenter en raison de taux plasmatiques de FVIII élevés de manière persistante.
L’effet secondaire suivant a été fréquemment observé (peut affecter jusqu’à 1 personne sur 10) :
· Réactions au point d’injection
Les effets secondaires suivants ont été observés peu fréquemment (peuvent affecter jusqu’à 1 personne sur 100) :
· Etourdissements
· Troubles ou diminution de la sensation tactile (Paresthésie, hypoesthésie)
· Bouffées de chaleur
· Démangeaisons
· Sensation d’oppression
· Frissons, sensation de froid
L’effet secondaire suivant a été observé avec une fréquence inconnue :
· Fièvre.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l'étiquette du flacon et la boîte. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas +25 °C. A conserver dans l'emballage extérieur, à l'abri de la lumière.
Ne pas congeler.
Pour des raisons de stérilité, il est recommandé d’utiliser le produit immédiatement après reconstitution. Toutefois, la stabilité physico-chimique en cours d'utilisation, a été démontrée pendant 24 heures à +25 °C.
N'utilisez pas ce médicament si vous constatez une solution trouble ou qui contient un dépôt.
Ne jetez aucun médicament au tout à l’égout ou avec les ordures ménagères. Demandez à votre pharmacien ou votre infirmier/ère d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient WILFACTIN 100 UI/mL, poudre et solvant pour solution injectable
· La substance active est : le Facteur von Willebrand humain* (500 UI, 1000 UI ou 2000 UI) exprimé en Unités Internationales du Cofacteur de la Ristocétine (VWF:RCo).
Après reconstitution avec 5 mL (500 UI), 10 mL (1000 UI) ou 20 mL (2000 UI) d'eau pour préparations injectables, un flacon contient 100 UI/mL de facteur von Willebrand humain.
Avant l'ajout d'albumine, l'activité spécifique est supérieure ou égale à 60 UI de VWF:RCo/mg de protéines.
· Les autres composants sont :
Poudre : albumine humaine, chlorhydrate d'arginine, glycine, citrate de sodium et chlorure de calcium dihydraté.
Solvant : eau pour préparations injectables.
WILFACTIN se présente sous la forme d'une poudre ou d’un solide friable de couleur blanche ou jaune pâle et d'un solvant clair ou incolore pour solution injectable après reconstitution avec un système de transfert.
WILFACTIN est disponible en présentations de 500 UI/5 mL, de 1000 UI/10 mL et de 2000 UI/20 mL.
La solution reconstituée doit être limpide ou légèrement opalescente, incolore ou légèrement jaune.
Titulaire de l’autorisation de mise sur le marché
3, AVENUE DES TROPIQUES
ZA DE COURTABŒUF
91940 LES ULIS
Exploitant de l’autorisation de mise sur le marché
3, AVENUE DES TROPIQUES
ZA DE COURTABŒUF
91940 LES ULIS
LFB-BIOMEDICAMENTS
3 AVENUE DES TROPIQUES
ZA DE COURTABOEUF
91940 LES ULIS
ou
LFB-BIOMEDICAMENTS
59 RUE TREVISE
BP 2006
59011 LILLE CEDEX
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen et au Royaume-Uni (Irlande du Nord) sous les noms suivants :
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
INSTRUCTION D’UTILISATION :
Posologie
Généralement, l’administration d’une UI/kg de facteur Willebrand augmente le taux plasmatique de VWF:RCo d'environ 0,02 UI/mL (2 %).
Des taux de VWF:RCo > 0,6 UI/mL (60 %) et de FVIII:C > 0,4 UI/mL (40 %) doivent être atteints.
L'hémostase est généralement assurée lorsque le facteur VIII coagulant (FVIII:C) atteint un taux de 0,4 UI/mL (40 %). L'injection de facteur Willebrand seul induit une remontée progressive du taux de FVIII:C qui n'atteint son maximum que dans un délai de 6 à 12 heures. Elle ne peut pas corriger immédiatement le taux de FVIII:C. Par conséquent, si le taux initial en FVIII:C du patient se situe
au-dessous du seuil critique, dans toutes les situations où une correction rapide de l'hémostase est nécessaire, tels que le traitement d'une hémorragie, un traumatisme sévère ou une intervention chirurgicale en urgence, il est nécessaire de co-administrer un facteur VIII associé au facteur von Willebrand, afin de parvenir à un taux de FVIII:C suffisant pour assurer l'hémostase.
Cependant, si une élévation immédiate du taux de FVIII:C n’est pas nécessaire, notamment en cas d’intervention chirurgicale programmée, ou si le taux de base de FVIII:C est suffisant pour assurer l’hémostase, le médecin peut décider de ne pas recourir à la co-administration de facteur VIII lors de la première injection de facteur von Willebrand.
· Première injection
La première dose est de 40 à 80 UI/kg de WilFACTIN pour le traitement des hémorragies ou d’un traumatisme, associée à la quantité nécessaire de facteur VIII, calculée selon le taux plasmatique de base de FVIII:C du patient, afin d’atteindre le taux approprié de FVIII:C, immédiatement avant l'intervention ou le plus tôt possible après la survenue de l'épisode hémorragique ou du traumatisme sévère. En cas de chirurgie, la première injection doit être administrée 1 h avant l'intervention.
Une dose initiale de 80 UI/kg de WILFACTIN peut être indiquée, en particulier chez les patients atteints du type 3 de la maladie de von Willebrand, pour lesquels des doses supérieures peuvent être nécessaires, afin de maintenir des taux suffisants.
En cas d'intervention programmée, un délai de 12 à 24 heures est recommandé entre la première injection de WILFACTIN et l'acte chirurgical qui sera précédé d'une seconde injection
pré-opératoire de WilFACTIN 1 heure avant l’intervention. Dans ce cas, il n'est pas nécessaire de co-administrer du facteur VIII, dans la mesure où le FVIII:C a atteint un taux critique de 0,4 UI/mL (40 %) avant l'intervention. Toutefois, ce taux doit être vérifié pour chaque patient.
· Injections suivantes
Le traitement sera poursuivi, si nécessaire, par WILFACTIN seul à la dose de 40 à 80 UI/kg par jour, en une ou deux injections quotidiennes, pendant un à plusieurs jours.
La dose et la fréquence des injections doivent toujours être adaptées au type de chirurgie, à l'état clinique et biologique du patient (VWF:RCo et FVIII:C) et au type et à la gravité de l'accident
· Prophylaxie à long terme
WILFACTIN peut être administré en prophylaxie à long terme à une posologie adaptée à chaque individu. Des posologies allant de 40 à 60 UI/kg de WILFACTIN, administrées 2 ou 3 fois par semaine, permettent de diminuer le nombre d'épisodes hémorragiques.
· Traitement ambulatoire
Le traitement à domicile peut être initié sur décision du praticien, notamment en cas de saignements mineurs à modérés ou lors d'une prophylaxie à long terme pour prévenir les saignements.
Population pédiatrique :
Pour chaque indication, la posologie dépend du poids corporel. La dose et la durée du traitement doivent être adaptées à l’état clinique du patient et à ses taux plasmatiques de VWF:RCo et de FVIII:C.
· Première injection :
o Chez les enfants âgés de moins de 6 ans, la dose initiale peut être déterminée en fonction de la récupération progressive (RP) du patient ; si les données de RP ne sont pas disponibles, une dose initiale de 60 à 100 UI/kg peut être nécessaire, dans l’objectif d’augmenter les taux de VWF:Rco jusqu’à 100 UI/dL.
o Chez les adolescents et les enfants âgés de plus de 6 ans, la posologie est la même que chez les patients adultes.
· Injections suivantes :
Chez les enfants et les adolescents, les doses suivantes doivent être calculées individuellement en fonction de l’état clinique et des taux de VWF:RCo, et ajustées en fonction de la réponse clinique.
En cas d’intervention chirurgicale programmée :
o Chez les enfants âgés de moins de 6 ans, la première dose sera administrée 12 à 24 heures avant la procédure et l’injection suivante pourra être administrée 30 minutes avant la procédure.
o Chez les adolescents et les enfants âgés de plus de 6 ans, la posologie est la même que chez les patients adultes.
· Prophylaxie :
Chez les enfants et les adolescents, la dose et la fréquence des nouvelles administrations doivent être calculées individuellement en fonction de la récupération progressive et des taux de VWF:RCo, et ajustées en fonction de la réponse clinique.
Méthode d'administration
Administration intraveineuse.
Reconstitution
Les règles d'asepsie en vigueur doivent être appliquées. Le système de transfert n'est utilisé que pour reconstituer le médicament, comme décrit ci-dessous. Il n'est pas destiné à l'administration du médicament au patient.
|
|
· Amener les deux flacons (poudre et solvant) à une température ne dépassant pas 25 °C.
· Retirer la capsule protectrice du flacon de solvant (eau pour préparations injectables) et du flacon de poudre.
· Désinfecter la surface de chaque bouchon.
· Retirer l’opercule du dispositif Mix2Vial. Sans extraire le dispositif de son emballage, enclencher l’extrémité bleue du Mix2Vial sur le bouchon du flacon de solvant.
· Retirer puis jeter l’emballage. Prendre soin de ne pas toucher la partie maintenant exposée du dispositif.
· Retourner l’ensemble flacon de solvant-dispositif et l’enclencher sur le flacon de poudre par la partie transparente du dispositif. Le solvant est transféré automatiquement dans le flacon de poudre. Maintenir l’ensemble et agiter doucement, d’un mouvement circulaire, pour dissoudre totalement le produit.
· En maintenant la partie produit reconstituée d’une main et la partie solvant de l’autre, séparer les flacons en dévissant le dispositif Mix2Vial.
|

La poudre est généralement dissoute instantanément et doit être dissoute en moins de 5 minutes.
La solution obtenue est limpide ou légèrement opalescente, incolore ou légèrement jaune.
Le produit reconstitué doit être inspecté visuellement pour vérifier la présence éventuelle de particules ou d’une décoloration avant administration.
Ne pas utiliser de solution présentant un trouble ou contenant un dépôt.
Ne pas mélanger à d’autres médicaments.
Ne pas diluer le produit reconstitué.
Administration
|
|
· Tenir le flacon du produit reconstitué verticalement en vissant une seringue stérile sur le dispositif Mix2Vial. Aspirer lentement le produit dans la seringue.
· Une fois le produit transféré dans la seringue, tenir celle-ci fermement (piston dirigé vers le bas), dévisser le dispositif Mix2Vial et le remplacer par une aiguille intraveineuse ou une aiguille épicrânienne.
· Expulser l’air de la seringue et piquer la veine après désinfection.
· Injecter lentement par voie intraveineuse en une seule fois, immédiatement après reconstitution, sans dépasser un débit de 4 ml/minute. |
Conservation après reconstitution
Pour des raisons de stérilité, il est recommandé d’utiliser le produit immédiatement après reconstitution. Toutefois, la stabilité physico-chimique en cours d’utilisation, a été démontrée pendant 24 heures à +25 °C.