Dernière mise à jour le 01/06/2026
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique : SOLUTIONS D'ELECTROLYTES - code ATC : B05XA03
B : sang et organes hématopoïétiques
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, est indiqué:
· pour rééquilibration ionique par apport de chlorure et de sodium ;
· pour le traitement de la déshydratation extracellulaire ;
· pour le traitement de l’hypovolémie ;
· comme véhicule ou diluant de médicaments compatibles pour administration parentérale.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 02/03/2016 | Inscription (CT) | Le service médical rendu par CHLORURE DE SODIUM AGUETTANT 0,9 POUR CENT, solution pour perfusion est important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 02/03/2016 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux présentations déjà inscrites. |
| V (Inexistant) | Avis du 05/03/2003 | Inscription (CT) | Absence d'amélioration du service médical rendu (ASMR V) par rapport aux autres solutions de chlorure de sodium à 0,9 pour cent. |
ANSM - Mis à jour le : 11/01/2019
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chlorure de sodium............................................................................................................... 0,90 g
pour 100 ml de solution pour perfusion
Un flacon de 50 mL contient 0,45 g de chlorure de sodium.
Un flacon de 100 mL contient 0,9 g de chlorure de sodium.
Un flacon de 125 mL contient 1,125 g de chlorure de sodium.
Un flacon de 250 mL contient 2,25 g de chlorure de sodium.
Un flacon de 500 mL contient 4,5 g de chlorure de sodium.
Un flacon de 1000 mL contient 9 g de chlorure de sodium.
Une poche de 2000 mL contient 18 g de chlorure de sodium.
Une poche de 3000 mL contient 27 g de chlorure de sodium.
Sodium (Na+): 154 mmol/L
Chlorures (Cl-): 154 mmol/L
Pour la liste complète des excipients, voir rubrique 6.1.
Solution limpide et incolore.
Osmolarité : 308 mOsm/L
pH compris entre 3,5 et 7
4.1. Indications thérapeutiques
· Rééquilibration ionique par apport de chlorure et de sodium ;
· Déshydratation extracellulaire ;
· Hypovolémie ;
· Véhicule ou diluant de médicaments compatibles pour administration parentérale.
4.2. Posologie et mode d'administration
Les doses peuvent être exprimées en mEq ou mmol de sodium, masse de sodium ou masse de sel de sodium (1 g NaCl = 394 mg, 17,1 mEq ou 17,1 mmol de Na et Cl).
La posologie, le débit et la durée d’administration doivent être déterminés en fonction de plusieurs facteurs comprenant l'âge, le poids, l'état clinique, le traitement concomitant et en particulier l'état d'hydratation du patient, des besoins en ions sodium et chlorure, la réponse clinique et biologique au traitement. L’équilibre hydrique et les concentrations en électrolytes sériques doivent être surveillés attentivement.
Pour le traitement de la déshydratation extracellulaire isotonique et de la déplétion sodique la posologie recommandée est :
· Adultes : 500 ml à 3 litres/24 h
· Population pédiatrique : 20 à 100 ml par 24 h et par kg de poids corporel. La posologie, le débit et le volume de perfusion dépendent de l'âge du poids, du statut clinique et métabolique du patient et du traitement concomitant. La solution doit être administrée par un personnel expérimenté.
Lorsque la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT est utilisée comme véhicule ou diluant pour préparation injectable d'autres médicaments, la posologie et le débit de perfusion seront principalement fonction de la nature et de la posologie du médicament à administrer.
Vitesse maximale de perfusion
La vitesse maximale de perfusion dépend de l'état clinique.
La plupart des cas de syndrome de démyélinisation osmotique rapportés sont survenus à la suite d’une vitesse de correction élevée (voir rubriques 4.4 et 4.9).
Mode d’administration
Perfusion intraveineuse, injection intramusculaire ou sous-cutanée.
Lorsque la solution est utilisée pour la dilution et l’administration de médicaments complémentaires, les instructions d'utilisation des substances ajoutées détermineront les volumes appropriés, les modes et voies d’administration pour chaque traitement.
Pour les précautions à prendre avant la manipulation ou l’administration du médicament, voir la rubrique 6.6.
· Hyperchlorémie
· Hypernatrémie
· Cas sévères d’inflation hydrique et de rétention hydro-sodée particulièrement en cas d’insuffisance cardiaque décompensée, d’insuffisance hépatique décompensée (insuffisance œdémato-ascitique des cirrhoses), de prééclampsie / éclampsie.
Les contre-indications relatives au(x) médicament(s) ajouté(s) doivent être prises en compte.
4.4. Mises en garde spéciales et précautions d'emploi
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, est une solution isotonique.
La rééquilibration en sodium ne doit pas être effectuée à un rythme trop rapide, en particulier en raison d’un risque de complications neurologiques graves, tel que le syndrome de démyélinisation osmotique (voir rubriques 4.2 et 4.9).
En fonction du volume et du débit de perfusion, l’administration intraveineuse de chlorure de sodium peut provoquer :
· une surcharge hydrique et/ou en soluté conduisant à une hyperhydratation/hypervolémie (ex., états congestifs, notamment œdème périphérique) ;
· des perturbations électrolytiques cliniquement significatives et un déséquilibre acido-basique.
Utiliser ce médicament avec précaution chez les patients atteints d'hypertension, d'insuffisance cardiaque, d'insuffisance hépatocellulaire avec œdème et ascite, de cirrhose du foie, d'œdème périphérique ou pulmonaire, de fonction rénale altérée, d’obstruction du tractus urinaire, d’acidose métabolique, de pré-éclampsie, d'hyperaldostéronisme, d’hypervolémie, d’hypoprotéinémie ou d'autres affections et traitements (ex., corticostéroïdes) associés à une rétention hydrosodée (voir rubrique 4.5).
Ce traitement doit être effectué sous surveillance médicale stricte, la posologie devant être adaptée selon les modifications hydroélectrolytiques, en particulier les ions sodium et chlorure. L’administration doit être réalisée sous surveillance régulière et attentive.
Extravasation
Le site du cathéter doit être régulièrement contrôlé pour détecter les signes d'extravasation. En cas d'extravasation, l'administration doit être interrompue immédiatement, tout en maintenant en place la canule ou le cathéter inséré pour une prise en charge immédiate du patient. Si possible, une aspiration doit être pratiquée à travers la canule/le cathéter inséré afin de réduire la quantité de liquide présent dans les tissus avant de retirer la canule/le cathéter. Si une extrémité est atteinte, le membre concerné doit être surélevé.
Selon le produit extravasé (y compris les produits mélangés avec CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, le cas échéant) et le stade/l'étendue des lésions éventuelles, des mesures spécifiques appropriées doivent être prises. Les options thérapeutiques peuvent inclure des interventions non pharmacologiques, pharmacologiques et/ou chirurgicales. En cas de dégradation de la zone affectée (douleur continue, nécrose, ulcération), un chirurgien plasticien doit être consulté immédiatement (voir rubrique 4.8).
Le site d'extravasation doit être contrôlé au moins toutes les 4 heures pendant les premières 24 heures, puis une fois par jour.
Risque d'embolie gazeuse (voir rubrique 6.6)
· Ne pas utiliser les poches plastiques pour des connexions en série. Cette utilisation pourrait entraîner une embolie gazeuse en raison de l'aspiration de l'air résiduel de la première poche avant la fin de l'administration de solution venant de la deuxième poche.
· L’exercice d’une pression sur le récipient en plastique flexible contenant la solution intraveineuse pour augmenter le débit peut entraîner une embolie gazeuse si l’air résiduel contenu dans le récipient n’est pas complètement évacué avant l’administration.
· L’utilisation d’un set d’administration par voie intraveineuse avec une prise d’air en position ouverte pourrait entraîner une embolie gazeuse. Les sets d’administration par voie intraveineuse avec une prise d’air en position ouverte ne doivent pas être utilisés avec des récipients en plastique souple.
Population gériatrique
Lors de la sélection du type de solution pour perfusion et du volume/débit de perfusion pour un patient âgé, il est nécessaire de prendre en considération la susceptibilité de ces patients à présenter des maladies cardiaques, rénales, hépatiques ou autres, ainsi que leurs traitements médicamenteux concomitants.
Population pédiatrique
Chez les nouveau-nés, les nourrissons et les enfants, l'administration du produit nécessite une surveillance accrue.
Chez le nouveau-né et le prématuré, il peut exister une rétention de sodium en excès due à une fonction rénale immature. Chez ces patients, les perfusions répétées de chlorure de sodium doivent être réalisées uniquement après la détermination des concentrations plasmatiques en sodium.
Le dispositif de perfusion intraveineuse et le matériel d'administration doivent être contrôlés régulièrement.
En cas d'ajout de médicament, vérifier la compatibilité, la limpidité et la couleur avant usage (voir rubrique 6.2).
Ne pas conserver le mélange (voir rubrique 6.6).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Associations faisant l'objet de précautions d’emploi
+ Lithium
L'administration de CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, peut entraîner une diminution des concentrations en lithium et donc un risque de baisse de l’efficacité du lithium. En effet, la clairance rénale du sodium et du lithium peut être augmentée lors de l'administration de CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion. La prudence est donc recommandée chez les patients qui ont un traitement au lithium.
+ Corticostéroïdes
Les corticostéroïdes sont associés à une rétention hydrosodée (avec œdème et hypertension). Voir rubrique 4.4.
4.6. Fertilité, grossesse et allaitement
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion peut être utilisé pendant la grossesse ou l'allaitement, si nécessaire.
Les risques et les bénéfices pour chaque patiente doivent être attentivement considérés avant d'administrer la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT.
La prudence est recommandée chez les patientes souffrant de pré-éclampsie (voir rubriques 4.3 et 4.4).
Lorsqu’un médicament est ajouté, la nature du médicament et son utilisation pendant la grossesse et l’allaitement doivent être évaluées séparément.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables pouvant survenir chez des patients traités avec CHLORURE DE SODIUM 0,9 % AGUETTANT par perfusion intraveineuse sont indiqués ci-dessous.
Les effets indésirables listés dans cette rubrique sont mentionnés selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 et < 1/10), peu fréquent (≥ 1/1000 et < 1/100), rare (≥ 1/10000 et < 1/1000), très rare (< 1/10000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classes de systèmes d’organes (SOC) |
Effets indésirables (Termes MedDRA) |
Fréquence |
|
Affections du système nerveux |
Tremblements |
Indéterminée |
|
Troubles du métabolisme et de la nutrition |
Hypervolémie Hypernatrémie Acidose métabolique hyperchlorémique |
Indéterminée |
|
Affections de la peau et du tissu sous-cutané |
Urticaire Eruption cutanée Prurit |
Indéterminée |
|
Affections vasculaires |
Thrombose veineuse Thrombophlébite Hypotension |
Indéterminée |
|
Troubles généraux et anomalies au site d’administration |
Frissons |
|
|
Fièvre |
||
|
Infection au niveau du site de perfusion |
||
|
Irritation au site de perfusion |
Indéterminée |
|
|
Extravasation |
||
|
Réaction locale |
||
|
Douleur localisée* Nécrose / ulcère* |
* Effets indésirables pouvant être notamment associés à une extravasation
Lorsque CHLORURE DE SODIUM 0,9% AGUETTANT est utilisé comme véhicule ou diluant pour des préparations injectables d'autres médicaments, d'autres effets indésirables associés au(x) médicament(s) ajouté(s) à la solution peuvent survenir.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Symptômes du surdosage en chlorure de sodium
Une administration excessive de CHLORURE DE SODIUM 0,9% AGUETTANT, solution pour perfusion, peut conduire à une hypernatrémie entraînant une déshydratation intracellulaire, qui doit être traitée en milieu spécialisé. Les effets indésirables généraux de l'excès de sodium comprennent nausées, vomissements, diarrhée, crampes abdominales, soif, diminution de la sécrétion de salive et de larmes, sudation, fièvre, tachycardie, hypertension, insuffisance rénale, œdème cérébral, œdème pulmonaire et périphérique, arrêt respiratoire, céphalées, étourdissement, impatiences, irritabilité, lipothymie, contraction et raideur musculaire, convulsions, coma et décès.
Les signes cliniques du syndrome de démyélinisation osmotique sont progressifs : confusion, dysarthrie, dysphagie, faiblesse des membres, puis tétraplégie, délire et finalement coma. Les symptômes cliniques surviennent plusieurs jours après une correction trop rapide et/ou trop importante de l’hyponatrémie (voir rubriques 4.2 et 4.4).
Les chlorures en excès dans l'organisme peuvent provoquer une perte de bicarbonate avec une acidose.
Traitement
La première mesure à prendre est la réduction de la vitesse de perfusion ou l'arrêt de la perfusion.
Le traitement consiste en la surveillance de la natrémie et en l'administration de solution pour perfusion de glucose. En cas de convulsions le diazépam pourra être administré.
Lorsque le CHLORURE DE SODIUM 0,9% AGUETTANT, solution pour perfusion, est utilisé comme véhicule ou diluant pour des préparations injectables d'autres médicaments, des signes et symptômes de perfusion excessive peuvent provenir du médicament ajouté. En cas de perfusion excessive accidentelle, interrompre le traitement et observer chez le patient toute apparition des signes et symptômes cliniques liés au médicament administré. Instaurer un traitement symptomatique et de soutien adapté, en fonction des besoins.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : SOLUTIONS D’ELECTROLYTES, code ATC : B05XA03 (B: sang et organes hématopoïétiques).
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, est une solution isotonique dont l'osmolarité est d’environ 308 mOsm/l.
Les propriétés pharmacodynamiques de la solution sont celles des ions sodium et chlorure, qui maintiennent l'équilibre hydroélectrolytique.
Les ions tels que le sodium circulent à travers la membrane cellulaire, en utilisant des mécanismes de transport variés, parmi lesquels la pompe à sodium (Na+, K+-ATPase). Le sodium joue un rôle important dans la neurotransmission et l'électrophysiologie cardiaque, ainsi que dans le métabolisme rénal.
En cas d’ajout de médicament, la pharmacodynamie de la préparation dépendra aussi du médicament ajouté.
5.2. Propriétés pharmacocinétiques
Étant donné que le chlorure de sodium est administré par voie intraveineuse, son absorption est complète, soit de 100 %.
Élimination
Les ions sodium et chlorure sont excrétés principalement dans les urines. De faibles quantités de sodium sont éliminées dans les fèces et la sueur.
En cas d’ajout de médicament, la pharmacocinétique de la préparation dépendra aussi du médicament ajouté.
5.3. Données de sécurité préclinique
Les données de sécurité de l'additif doivent être considérées séparément.
Eau pour préparations injectables.
L'incompatibilité du médicament vis-à-vis de la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT doit être déterminée en contrôlant un éventuel changement de couleur et/ou une éventuelle formation de précipité, de complexe insoluble ou de cristaux. Se référer également à la notice accompagnant le médicament à ajouter.
En cas d'ajout de médicament, vérifier si le médicament est compatible avec la zone de pH de la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT.
Lorsqu’un médicament compatible est ajouté à la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT, le mélange doit être administré immédiatement.
Les médicaments connus pour être incompatibles ne doivent pas être utilisés.
Flacon (verre) : 5 ans.
Poche (PVC plastifié) : 18 mois.
Durée de conservation lors de l'utilisation :
Après ouverture/dilution, le produit doit être utilisé immédiatement.
6.4. Précautions particulières de conservation
Flacon (verre) : Ce médicament ne nécessite pas de précautions particulières de conservation.
Poche (PVC plastifié) : à conserver à une température ne dépassant pas 25° C.
6.5. Nature et contenu de l'emballage extérieur
Flacon de 125 ml, 250 ml, 500 ou 1000 ml en verre incolore de type II, fermé par un bouchon en caoutchouc chlorobutyl.
Poche souple en chlorobutyle de vinyle (PVC) plastifié de 50 ml, 10 ml, 250 ml, 500 ml, 1000 ml 2000 ml ou 3000 ml munie de deux tubes d’accès en PVC plastifié :
· avec embout Double Accès Trocardable (DAT) comprenant un site d’injection en polycarbonate cristal et polyisoprène de synthèse et un site de connexion en polycarbonate cristal et caoutchouc chlorobutyle.
· Ou embout T-OFF comprenant un site d’injection droit en polycarbonate cristal et polyisoprène de synthèse ainsi qu’un site de connexion trocardable en PVC.
· Ou embout Luer Site (LS) comprenant un site de connexion en polycarbonate blanc et un site d’injection en polycarbonate blanc et polyisoprène de synthèse.
· Ou embout Luer Accès (LA) comprenant un site d’injection en polycarbonate cristal et polyisoprène de synthèse et un site de connexion en polycarbonate blanc.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Ne pas utiliser si l'emballage / la poche / le flacon est endommagé(e).
Ne pas réutiliser : usage unique.
Eliminer toute poche/tout flacon partiellement utilisé(e).
Ne pas reconnecter une poche partiellement utilisée.
Retirer le suremballage de la poche juste avant utilisation. Le conditionnement primaire maintient la stérilité du produit.
En cas d’ajout de médicament, bien mélanger la solution avant utilisation.
La solution doit être inspectée visuellement afin de détecter toute particule, tout dommage de la poche/du flacon et tout signe visible de détérioration avant administration.
En cas d’ajout de médicament, la solution doit être administrée avec un matériel stérile et en utilisant une technique aseptique. Le matériel doit être amorcé à l'aide de la solution pour éviter toute introduction d'air dans le système.
Risque d'embolie gazeuse
· Ne pas utiliser les poches plastiques pour des connexions en série. Cette utilisation pourrait entraîner une embolie gazeuse en raison de l'aspiration de l'air résiduel de la première poche avant la fin de l'administration de solution venant de la deuxième poche.
· L’exercice d’une pression sur le récipient en plastique flexible contenant la solution intraveineuse pour augmenter le débit peut entraîner une embolie gazeuse si l’air résiduel contenu dans le récipient n’est pas complètement évacué avant l’administration.
· L’utilisation d’un set d’administration par voie intraveineuse avec une prise d’air en position ouverte pourrait entraîner une embolie gazeuse. Les sets d’administration par voie intraveineuse avec une prise d’air en position ouverte ne doivent pas être utilisés avec des récipients en plastique souple.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
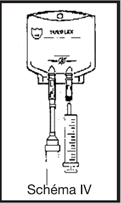
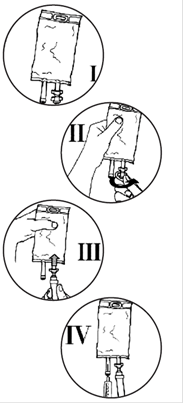
Mode d’emploi de la poche TULIFLEX DAT double accès trocardable
· vérifier l’intégrité du suremballage ;
· sortir la poche du suremballage et faire les vérifications habituelles (limpidité, absence de fuites, volume, nature de la solution, péremption)
* La présence de gouttelettes entre la poche et le suremballage est liée au procédé de stérilisation vapeur.
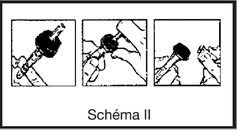
· ferme la prise d’air du perfuseur s’il y en a une et fermer le clamp
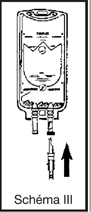
· casser l’obturateur de la tulipe pour libérer le site de perfusion stérile
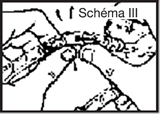
· perforer le site de perfusion en enfonçant le perforateur du perfuseur à l’intérieur du cône de la tulipe
· ouvrir le clam du perfuseur et purger. Fermer le clamp
· en cas d’ajouts de médicaments dans la poche : injecter le produit à ajouter en perforant le site d’injection de la poche à l’aide de l’aiguille de la seringue ou du dispositif de transfert.
· Agiter la poche pour homogénéiser la solution, le produit est prêt à être administré.
Mode d’emploi de la pocheTuliflex T-OFF
· vérifier l’intégrité du suremballage ;
· sortir la poche du suremballage et faire les vérifications habituelles (limpidité, absence de fuites, volume, nature de la solution, péremption)
* La présence de gouttelettes entre la poche et le suremballage est liée au procédé de stérilisation vapeur.
· fermer la prise d’air du perfuseur s’il y en a une et fermer le clamp
· ouvrir aseptiquement par torsion du twist-off
· perforer le site de perfusion en enfonçant le trocart du perfuseur avec un mouvement de rotation
· ouvrir le clamp du perfuseur et purger. Fermer le clamp
· en cas d’ajouts de médicaments dans la poche : injecter le produit à ajouter en perforant le site d’injection de la poche à l’aide de l’aiguille de la seringue ou du dispositif de transfert.
· Agiter la poche pour homogénéiser la solution, le produit est prêt à être administré.
Mode d’emploi de la poche TULIFLEX LA (Luer Accès)
· vérifier l’intégralité du suremballage
· sortir la poche du suremballage et faire les vérifications habituelles (limpidité, absence de fuites, nature de la solution, péremption)
* La présence de gouttelettes entre la poche et le suremballage est liée au procédé de stérilisation vapeur
· fermer le clamp du perfuseur
· connecter
· casser le rupteur
· ouvrir le clamp du perfuseur et purger. Fermer le clamp
· en cas d’ajouts de médicaments dans la poche : injecter le produit à ajouter en perforant le site d’injection de la poche à l’aide de l’aiguille de la seringue ou du dispositif de transfert.
· Agiter la poche pour homogénéiser la solution, le produit est prêt à être administré.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1, RUE ALEXANDER FLEMING
69007 LYON
FRANCE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 340 194 6 3 : 50 ml en flacon de 125 ml (verre incolore), boîte de 1
· 34009 553 808 1 8 : 50 ml en flacon de 125 ml (verre incolore), boîte de 24
· 34009 340 191 7 3 : 100 ml en flacon de 125 ml (verre incolore), boîte de 1
· 34009 553 809 8 6 : 100 ml en flacon de 125 ml (verre incolore), boîte de 24
· 34009 340 192 3 4 : 125 ml en flacon (verre incolore), boîte de 1
· 34009 555 090 0 4 : 125 ml en flacon (verre incolore), boîte de 24
· 34009 557 046 9 0 : 125 ml en flacon de 250 ml (verre incolore), boîte de 12
· 34009 318 438 3 2 : 250 ml en flacon (verre incolore), boîte de 1
· 34009 557 047 5 1 : 250 ml en flacon (verre incolore), boîte de 12
· 34009 318 440 8 2 : 500 ml en flacon (verre incolore), boîte de 1
· 34009 557 048 1 2 : 500 ml en flacon (verre incolore), boîte de 6
· 34009 318 441 4 3 : 1000 ml en flacon (verre incolore), boîte de 1
· 34009 557 049 8 0 : 1000 ml en flacon (verre incolore), boîte de 6
· 34009 340 180 5 3 : 2000 ml en poche (PVC plastifié) avec embout DAT, boîte de 1
· 34009 555 096 9 1 : 2000 ml en poche (PVC plastifié) avec embout DAT, boîte de 5
· 34009 340 181 1 4 : 3000 ml en poche (PVC plastifié) avec embout DAT, boîte de 1
· 34009 554 405 8 1 : 3000 ml en poche (PVC plastifié) avec embout DAT, boîte de 5
· 34009 562 073 0 5 : 3000 ml en poche (PVC plastifié) avec embout DAT, boîte de 4
· 34009 367 646 5 1 : 2000 ml en poche (PVC plastifié) avec embout T-OFF, boîte de 1
· 34009 565 870 9 4 : 2000 ml en poche (PVC plastifié) avec embout T-OFF, boîte de 5
· 34009 367 647 1 2 : 3000 ml en poche (PVC plastifié) avec embout T-OFF, boîte de 1
· 34009 565 871 5 5 : 3000 ml en poche (PVC plastifié) avec embout T-OFF, boîte de 4
· 34009 554 728 1 0 : 2000 ml en poche (PVC plastifié) avec embout LS, boîte de 5
· 34009 557 041 7 1 : 3000 ml en poche (PVC plastifié) avec embout LS, boîte de 4
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 11/01/2019
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion
Chlorure de sodium
Vous devez toujours utiliser ce médicament en suivant scrupuleusement les informations fournies dans cette notice ou par votre médecin, votre pharmacien ou votre infirmier/ère.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Adressez-vous à votre pharmacien pour tout conseil ou information.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
· Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien.
2. Quelles sont les informations à connaître avant d'utiliser CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion ?
3. Comment utiliser CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : SOLUTIONS D'ELECTROLYTES - code ATC : B05XA03
B : sang et organes hématopoïétiques
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, est indiqué:
· pour rééquilibration ionique par apport de chlorure et de sodium ;
· pour le traitement de la déshydratation extracellulaire ;
· pour le traitement de l’hypovolémie ;
· comme véhicule ou diluant de médicaments compatibles pour administration parentérale.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion ?
N’utilisez jamais CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion :
· si vous avez un taux de chlorure anormalement élevé dans le sang (hyperchlorémie) ;
· si vous avez un taux de sodium anormalement élevé dans le sang (hypernatrémie) ;
· si vous souffrez d’une rétention sévère d’eau (inflation hydrique) et/ou de sodium, en particulier en cas :
o de défaillance des fonctions du cœur (insuffisance cardiaque) ;
o de défaillance des fonctions du foie non contrôlée (insuffisance hépatique décompensée, insuffisance oedémato-ascitique des cirrhoses) ;
o de poussée d'hypertension artérielle au cours de la grossesse (pré-éclampsie), convulsions au cours de la grossesse, associée à une poussée d'hypertension artérielle (éclampsie).
De plus, lorsque la solution de chlorure de sodium est utilisée comme véhicule ou diluant, les contre-indications relatives au(x) médicament(s) ajouté(s) doivent être prises en compte.
EN CAS DE DOUTE IL EST INDISPENSABLE DE DEMANDER L’AVIS DE VOTRE MEDECIN OU DE VOTRE PHARMACIEN.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion.
L’administration de ce médicament doit se faire sous surveillance médicale.
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, est une solution isotonique (dont la concentration est la même que celle du plasma sanguin).
La rééquilibration en sodium ne doit pas être effectuée à un rythme trop rapide.
Des signes d’irritation veineuse ou d’inflammation de la paroi d’une veine peuvent apparaître au niveau du site de perfusion.
Informez votre médecin si vous souffrez ou avez souffert des troubles suivants :
· une pression artérielle élevée (hypertension),
· une défaillance des fonctions du cœur (insuffisance cardiaque),
· une défaillance des fonctions du foie (insuffisance hépatocellulaire) avec présence anormale de liquide dans le ventre (ascite) et œdème,
· une accumulation de liquide sous-cutanée en particulier au niveau des chevilles (œdème périphérique),
· une accumulation de liquide au niveau des poumons (œdème pulmonaire),
· une maladie du foie (cirrhose),
· une fonction rénale altérée,
· une acidité élevée du sang (acidose métabolique),
· une poussée d'hypertension artérielle au cours de la grossesse (pré-éclampsie),
· une production anormalement élevée d’une hormone appelée l'aldostérone (hyperaldostéronisme),
· une augmentation du volume du sang (hypervolémie),
· une obstruction des voies urinaires,
· une diminution anormale du taux de protéines dans le sang (hypoprotéinémie) ou d'autres affections et traitements (ex., les corticostéroïdes) associés à une rétention d’eau et de sodium (voir rubrique « Autres médicaments et CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion » ci-dessous).
Lors d’une perfusion, votre médecin demandera des prélèvements d’échantillons de sang et d’urines pour surveiller :
· les quantités de fluides dans votre corps (équilibre hydrique),
· les concentrations en substances chimiques dans votre sang comme le sodium et le chlorure (électrolytes plasmatiques).
Vos signes vitaux seront également surveillés.
Chez les personnes âgées, l'administration du produit nécessite une surveillance accrue.
Enfants et adolescents
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, doit être administré avec précaution. Le débit et le volume de perfusion dépendent de l’âge, du poids, de l’état clinique et des paramètres métaboliques de l’enfant.
Chez le nouveau-né et le prématuré, il peut exister une rétention de sodium en excès due à une fonction rénale immature. Chez ces patients les perfusions répétées de chlorure de sodium doivent être réalisées uniquement après la détermination du taux de sodium dans le sang.
Autres médicaments et CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Il est particulièrement important que vous informiez votre médecin si vous prenez :
· des corticostéroïdes (médicaments anti-inflammatoires). Ces médicaments peuvent avoir pour conséquence l’accumulation de sel et d’eau dans votre corps (rétention hydrosodée), conduisant à un gonflement des tissus (œdème) et à une pression sanguine élevée (hypertension).
· du lithium (utilisé pour soigner les maladies psychiatriques). L’administration de chlorure de sodium peut entraîner une diminution du taux de lithium dans votre sang et donc un risque de baisse d’efficacité du lithium.
En cas d’ajout de médicament, votre médecin vérifiera sa compatibilité avec la solution avant que ce médicament ne vous soit administré.
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion avec des aliments et boissons
Sans objet.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Compte tenu des données disponibles, l'utilisation chez la femme enceinte ou qui allaite est possible.
La prudence est recommandée chez les patientes souffrant de pré-éclampsie (poussée d'hypertension artérielle au cours de la grossesse). Voir les rubriques « N'utilisez jamais CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion » et « Avertissements et précautions ».
Lorsqu’un médicament est ajouté, la nature du médicament et son utilisation pendant la grossesse et l’allaitement doivent être évaluées séparément.
Conduite de véhicules et utilisation de machines
Il n’existe pas d’information sur les effets du CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, sur l'aptitude à conduire des véhicules et à utiliser des machines.
CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion contient
Sans objet.
3. COMMENT UTILISER CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion ?
Votre médecin veillera à un débit de perfusion adapté à vos besoins et à votre état clinique.
SE CONFORMER A L'AVIS MEDICAL.
Mode et voie d'administration
Perfusion intraveineuse, injection intramusculaire ou sous-cutanée.
SE CONFORMER A L'AVIS MEDICAL.
Fréquence d'administration
SE CONFORMER A L'AVIS MEDICAL.
Durée du traitement
SE CONFORMER A L'AVIS MEDICAL
Si vous avez utilisé plus de CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion que vous n’auriez dû
Si vous avez reçu du CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, en excès, les symptômes suivants, associés à des taux de sodium anormalement élevés dans le sang (hypernatrémie), peuvent survenir :
· nausées (sensation de malaise),
· vomissements,
· diarrhée (selles molles),
· déshydratation,
· crampes abdominales,
· soif,
· sécheresse de la bouche,
· sécheresse des yeux,
· transpiration,
· fièvre,
· rythme cardiaque rapide (tachycardie),
· pression artérielle élevée (hypertension),
· insuffisance rénale,
· accumulation de liquide dans les poumons rendant la respiration difficile (œdème pulmonaire),
· accumulation de liquide sous-cutanée et en particulier au niveau des chevilles (œdème périphérique),
· arrêt respiratoire,
· maux de tête,
· étourdissement,
· impatiences (agitation),
· irritabilité,
· faiblesse (lipothymie),
· contraction et raideur musculaires,
· convulsions, coma, gonflement du cerveau (œdème cérébral) et mort.
Un taux de chlorure anormalement élevé dans le sang (hyperchlorémie) peut provoquer une acidification de votre sang (acidose) provoquant une fatigue, une confusion, une léthargie et un rythme respiratoire augmenté.
Une perfusion trop rapide et/ou trop importante de chlorure de sodium peut entraîner des troubles liés à l’apparition de lésions au niveau de vos nerfs, constituant ce que l’on appelle un « syndrome de démyélinisation osmotique » et se manifestant progressivement par une confusion, des troubles de la prononciation, une difficulté à avaler, une faiblesse des membres, puis une tétraplégie, un délire et finalement un coma.
Si vous développez l’un de ces symptômes, vous devez le signaler immédiatement à votre médecin ou à votre infirmier/ère. Votre perfusion sera interrompue ou sa vitesse réduite et vous recevrez un traitement adapté selon vos symptômes.
Lorsque le CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion, est utilisé comme véhicule ou diluant pour des préparations injectables d'autres médicaments, les signes et symptômes d’une perfusion excessive peuvent provenir du médicament ajouté.
Si vous oubliez d’utiliser CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion
Si vous arrêtez d’utiliser CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion
Votre médecin décidera quand arrêter la perfusion.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Fréquence indéterminée : ne peut être estimée sur la base des données disponibles · Tremblements · Fièvre · Frissons · Infection au niveau du site de perfusion · Réaction locale · Douleur locale · Urticaire · Eruption cutanée · Démangeaisons (prurit) · Irritation veineuse · Thrombose veineuse ou phlébite s'étendant à partir du site de perfusion (formation d’un caillot qui obstrue un vaisseau sanguin) · Baisse de la tension artérielle (hypotension) · Extravasation (passage du produit hors des vaisseaux) pouvant entraîner notamment une douleur locale, voire un ulcère ou la mort des cellules tissulaires (nécrose de la peau) · Hypervolémie (augmentation du volume du sang) · Hypernatrémie (un taux anormalement élevé de sel dans le sang) · Acidose métabolique hyperchlorémique (acidification du sang, associée à un taux anormalement élevé de chlorure dans le sang). Ces effets indésirables peuvent être associés à la technique d'administration ou à un débit d’administration trop rapide. Si un médicament a été ajouté à la solution pour perfusion, celui-ci peut aussi provoquer des effets indésirables. Consultez la notice de ce médicament pour identifier des effets indésirables possibles. Déclaration des effets secondaires Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.ansm.sante.fr En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants. N’utilisez pas ce médicament après la date de péremption indiquée sur la boite après EXP. La date de péremption fait référence au dernier jour de ce mois. Avant ouverture : Flacon : Ce médicament ne nécessite pas de précautions particulières de conservation. Poches : A conserver à une température ne dépassant pas +25°C. Durée de conservation lors de l'utilisation : Après ouverture/dilution, le produit doit être utilisé immédiatement. La solution doit être inspectée visuellement afin de détecter toute particule, tout dommage du flacon/de la poche et tout signe visible de détérioration avant administration. Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion · La substance active est : Chlorure de sodium........................................................................................................... 0,9 g Pour 100 mL de solution pour perfusion. Un flacon de 50 mL contient 0,45 g de chlorure de sodium. Un flacon de 100 mL contient 0,9 g de chlorure de sodium. Un flacon de 125 mL contient 1,125 g de chlorure de sodium. Un flacon de 250 mL contient 2,25 g de chlorure de sodium. Un flacon de 500 mL contient 4,5 g de chlorure de sodium. Un flacon de 1000 mL contient 9 g de chlorure de sodium. Une poche de 2000 mL contient 18 g de chlorure de sodium. Une poche de 3000 mL contient 27 g de chlorure de sodium. Sodium (Na+) : 154 mmol/L Chlorures (Cl-) : 154 mmol/L Osmolarité: 308 mOsm/L pH = 3,5 à pH = 7,0 · L’autre composant est : Eau pour préparations injectables. Ce médicament se présente sous forme de solution pour perfusion. Solution limpide et incolore. CHLORURE DE SODIUM 0,9 % AGUETTANT, solution pour perfusion est présentée en flacon (verre) de 50 ml, 100 ml, 125 ml, 250 ml, 500 ou 1000 ml et en poche (PVC plastifié) de 2000 ml ou 3000 ml. Toutes les présentations peuvent ne pas être commercialisées. Titulaire de l’autorisation de mise sur le marché 1, RUE ALEXANDER FLEMING 69007 LYON FRANCE Exploitant de l’autorisation de mise sur le marché 1, RUE ALEXANDER FLEMING 69007 LYON FRANCE LIEU-DIT CHANTECAILLE 07340 CHAMPAGNE FRANCE Noms du médicament dans les Etats membres de l'Espace Economique Européen Sans objet. La dernière date à laquelle cette notice a été révisée est : [à compléter ultérieurement par le titulaire] < {MM/AAAA}>< {mois AAAA}.> Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France). Les informations suivantes sont destinées exclusivement aux professionnels de santé: A. POSOLOGIE ET MODE D'ADMINISTRATION Les doses peuvent être exprimées en mEq ou mmol de sodium, masse de sodium ou masse de sel de sodium (1 g NaCl = 394 mg de Na, 17,1 mEq ou 17,1 mmol de Na et Cl). La posologie, le débit et la durée d’administration doivent être déterminés en fonction de plusieurs facteurs comprenant l'âge, le poids, l'état clinique, le traitement concomitant et en particulier l'état d'hydratation du patient, des besoins en ions sodium et chlorure, la réponse clinique et biologique au traitement. L’équilibre hydrique et les concentrations en électrolytes sériques doivent être surveillés attentivement. Pour le traitement de la déshydratation extracellulaire isotonique et de la déplétion sodique la posologie recommandée est: · Adultes : 500 ml à 3000 ml /24 h. · Population pédiatrique : 20 à 100 ml par 24 h et par kg de poids corporel. La posologie, le débit et le volume de perfusion dépendent de l’âge, du poids, du statut clinique et métabolique du patient et du traitement concomitant. La solution doit être administrée par un personnel expérimenté. Lorsque la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT est utilisée comme véhicule ou diluant pour préparation injectable d’autres médicaments, la posologie et le débit de perfusion seront principalement fonction de la nature et de la posologie du médicament à administrer. Vitesse maximale de perfusion La vitesse maximale de perfusion dépend de l'état clinique. La plupart des cas de syndrome de démyélinisation osmotique rapportés sont survenus à la suite d’une vitesse de correction élevée (voir rubriques 4.4 et 4.9). Mode d'administration : Perfusion intraveineuse, injection IM ou SC. Lorsque la solution est utilisée pour la dilution et l’administration de médicaments complémentaires, les instructions d'utilisation des substances ajoutées détermineront les volumes appropriés, les modes et voies d’administration pour chaque traitement. Pour les précautions à prendre avant la manipulation ou l’administration du médicament, voir la rubrique D. Extravasation Le site du cathéter doit être régulièrement contrôlé pour détecter les signes d'extravasation. En cas d'extravasation, l'administration doit être interrompue immédiatement, tout en maintenant en place la canule ou le cathéter inséré pour une prise en charge immédiate du patient. Si possible, une aspiration doit être pratiquée à travers la canule/le cathéter inséré afin de réduire la quantité de liquide présent dans les tissus avant de retirer la canule/le cathéter. Si une extrémité est atteinte, le membre concerné doit être surélevé. Selon le produit extravasé (y compris les produits mélangés avec CHLORURE DE SODIUM 0,9 % AGUETTANT, le cas échéant) et le stade/l'étendue des lésions éventuelles, des mesures spécifiques appropriées doivent être prises. Les options thérapeutiques peuvent inclure des interventions non pharmacologiques, pharmacologiques et/ou chirurgicales. En cas de dégradation de la zone affectée (douleur continue, nécrose, ulcération), un chirurgien plasticien doit être consulté immédiatement. Le site d'extravasation doit être contrôlé au moins toutes les 4 heures pendant les premières 24 heures, puis une fois par jour. B. INCOMPATIBILITES Comme avec toutes les solutions parentérales, la compatibilité des additifs avec la solution doit être vérifiée avant adjonction de médicament. L'incompatibilité du médicament vis-à-vis de la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT doit être déterminée en contrôlant un éventuel changement de couleur et/ou une éventuelle formation de précipité, de complexe insoluble ou de cristaux. Se référer également à la notice accompagnant le médicament à ajouter. En cas d'ajout de médicament, vérifier si le médicament est compatible avec la zone de pH de la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT. Lorsqu’un médicament compatible est ajouté à la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT, le mélange doit être administré immédiatement. Les médicaments connus pour être incompatibles ne doivent pas être utilisés. C. PRECAUTIONS PARTICULIERES D’ELIMINATION ET DE MANIPULATION Ne pas utiliser si l'emballage / la poche / le flacon est endommagé(e). Ne pas réutiliser : usage unique. Eliminer toute poche/tout flacon partiellement utilisé(e). Ne pas reconnecter une poche partiellement utilisée. Retirer le suremballage de la poche juste avant utilisation. Le conditionnement primaire maintient la stérilité du produit. En cas d’ajout de médicament, bien mélanger la solution avant utilisation. La solution doit être inspectée visuellement afin de détecter toute particule, tout dommage de la poche/du flacon et tout signe visible de détérioration avant administration. En cas d’ajout de médicament, la solution doit être administrée avec un matériel stérile et en utilisant une technique aseptique. Le matériel doit être amorcé à l'aide de la solution pour éviter toute introduction d'air dans le système. Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur. Risque d'embolie gazeuse · Ne pas utiliser les poches plastiques pour des connexions en série. Cette utilisation pourrait entraîner une embolie gazeuse en raison de l'aspiration de l'air résiduel de la première poche avant la fin de l'administration de solution venant de la deuxième poche. · L’exercice d’une pression sur le récipient en plastique flexible contenant la solution intraveineuse pour augmenter le débit peut entraîner une embolie gazeuse si l’air résiduel contenu dans le récipient n’est pas complètement évacué avant l’administration. · L’utilisation d’un set d’administration par voie intraveineuse avec une prise d’air en position ouverte pourrait entraîner une embolie gazeuse. Les sets d’administration par voie intraveineuse avec une prise d’air en position ouverte ne doivent pas être utilisés avec des récipients en plastique souple. Mode d'emploi pour la poche TULIFLEX DAT double accès trocardable Schéma n° I · vérifier l'intégrité du suremballage; · sortir la poche du suremballage et faire les vérifications habituelles (limpidité, absence de fuites*, volume, nature de la solution, péremption). * La présence de gouttelettes entre la poche et le suremballage est liée au procédé de stérilisation vapeur. Schéma n° II TULIFLEX DAT (double accès trocardable) Protocole d'utilisation · fermer la prise d'air du perfuseur s'il y en a une et fermer le clamp; · casser l'obturateur de la tulipe pour libérer le site de perfusion stérile. Schéma n° III · perforer le site de perfusion en enfonçant le perforateur du perfuseur à l'intérieur du cône de la tulipe; · ouvrir le clamp du perfuseur et purger. Fermer le clamp. Schéma n° IV · en cas d'ajouts de médicaments dans la poche: injecter le produit à ajouter en perforant le site d'injection de la poche à l'aide de l'aiguille de la seringue ou du dispositif de transfert. · agiter la poche pour homogénéiser la solution. Le produit est prêt à être administré. Mode d'emploi pour la poche TULIFLEX LA (Luer Accès) Schéma n° I · vérifier l'intégrité du suremballage; · sortir la poche du suremballage et faire les vérifications habituelles (limpidité, absence de fuites*, volume, nature de la solution, péremption). * La présence de gouttelettes entre la poche et le suremballage est liée au procédé de stérilisation vapeur. Schéma n° II · fermer le clamp du perfuseur · connecter. Schéma n° III · casser le rupteur; · ouvrir le clamp du perfuseur et purger. Fermer le clamp. Schéma n° IV · en cas d'ajouts de médicaments dans la poche: injecter le produit à ajouter en perforant le site d'injection de la poche à l'aide de l'aiguille de la seringue ou du dispositif de transfert; · agiter la poche pour homogénéiser la solution. Le produit est prêt à être administré. Mode d'emploi pour la poche TULIFLEX T-OFF · vérifier l'intégrité du suremballage; · sortir la poche du suremballage et faire les vérifications habituelles (limpidité, absence de fuites*, volume, nature de la solution, péremption). * La présence de gouttelettes entre la poche et le suremballage est liée au procédé de stérilisation vapeur. · fermer la prise d'air du perfuseur s'il y en a une et fermer le clamp; · ouvrir aseptiquement par torsion du twist-off; · perforer le site de perfusion en enfonçant le trocart du perfuseur avec un mouvement de rotation; · ouvrir le clamp du perfuseur et purger. Fermer le clamp; · en cas d'ajouts de médicaments dans la poche: injecter le produit à ajouter en perforant le site d'injection de la poche à l'aide de l'aiguille de la seringue ou du dispositif de transfert; · agiter la poche pour homogénéiser la solution. Le produit est prêt à être administré. D. DUREE DE CONSERVATION APRES RECONSTITUTION (ADDITIFS) La stabilité physico-chimique de tout médicament ajouté à de la solution de CHLORURE DE SODIUM 0,9 % AGUETTANT doit être vérifiée avant son utilisation. Après ouverture/dilution, le produit doit être utilisé immédiatement.