Base de données publique
des médicaments
Visiter [medicaments.gouv.fr] ![Visiter [medicaments.gouv.fr]](/img/icone_lien.png "Visiter [medicaments.gouv.fr] - nouvelle fenêtre")



LEVOCETIRIZINE RANBAXY 5 mg, comprimé pelliculé - Résumé des caractéristiques du produit |
 |
|
ANSM - Mis à jour le : 31/08/2018
LEVOCETIRIZINE RANBAXY 5 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE 
Chaque comprimé pelliculé contient 5 mg de dichlorhydrate de lévocétirizine.
Excipient à effet notoire : un comprimé contient 70,00 mg de lactose.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé.
Comprimé pelliculé blanc à blanc cassé, ovale, biconvexe avec "5" gravé sur une face, et l'autre face lisse.
4.1. Indications thérapeutiques
Traitement symptomatique de la rhinite allergique (incluant la rhinite allergique persistante) et de l'urticaire.
4.2. Posologie et mode d'administration
Chez les enfants de 2 à 6 ans, la formulation en comprimé pelliculé ne permet pas d’ajustement de la posologie. Il est recommandé d'utiliser une formulation pédiatrique de lévocétirizine.
Etant donné le manque de données pour cette population, l'administration de lévocétirizine aux enfants de moins de 2 ans est déconseillée.
Posologie
Adultes et adolescents à partir de 12 ans :
La posologie quotidienne recommandée est de 5 mg (1 comprimé pelliculé).
Patients âgés :
Il est recommandé d’ajuster la posologie chez les patients âgés souffrant d'insuffisance rénale modérée à sévère (voir Patients atteints d’insuffisance rénale ci-dessous).
Enfants de 6 à 12 ans :
La posologie quotidienne recommandée est de 5 mg (1 comprimé pelliculé).
Patients adultes atteints d’insuffisance rénale :
L'intervalle séparant les prises doit être ajusté en fonction de la fonction rénale. Se référer au tableau ci-dessous et adapter la posologie de la façon indiquée. Il est nécessaire de disposer d’une valeur de la clairance de la créatinine (ClCr) du patient exprimée en ml/min pour utiliser ce tableau posologique. La ClCrr (ml/min) peut être estimée à partir de la créatininémie (mg/dl) grâce à la formule suivante :
![]()
Adaptations posologiques pour les patients atteints d’insuffisance rénale :
|
Groupe |
Clairance de la créatinine |
Posologie et fréquence |
|
Fonction rénale normale |
≥80 |
5 mg une fois par jour |
|
Insuffisance rénale légère |
50–79 |
5 mg une fois par jour |
|
Insuffisance rénale modérée |
30–49 |
5 mg une fois tous les 2 jours |
|
Insuffisance rénale sévère |
<30 |
5 mg une fois tous les 3 jours |
|
Insuffisance rénale terminale – Patients sous dialyse |
<10 |
Contre-indiqué |
Chez les patients pédiatriques atteints d’insuffisance rénale, la posologie devra être ajustée individuellement en tenant compte de la clairance rénale du patient et de son poids corporel. On ne dispose d’aucune donnée spécifique sur les enfants atteints d’insuffisance rénale.
Patients atteints d’insuffisance hépatique :
Aucun ajustement de la posologie n'est nécessaire chez les patients atteints d’insuffisance hépatique isolée. Chez les patients souffrant conjointement d’insuffisance hépatique et d’insuffisance rénale, un ajustement de la posologie est recommandé (voir Patients atteints d’insuffisance rénale ci-dessus).
Durée de traitement :
La rhinite allergique intermittente (définie par la présence de symptômes moins de 4 jours par semaine et pendant moins de quatre semaines) sera traitée en fonction de la pathologie et de son historique ; le traitement peut être arrêté avec la disparition des symptômes et repris à leur réapparition des symptômes. En cas de rhinite allergique persistante (définie par la survenue de symptômes plus de 4 jours par semaine et pendant plus de 4 semaines), un traitement continu peut être proposé au patient pendant la période d'exposition aux allergènes.
L'expérience clinique acquise est de 6 mois de traitement avec 1 comprimé à 5 mg de lévocétirizine par jour.
Avec la cétirizine (forme racémique), il existe une expérience clinique allant jusqu'à un an de traitement pour l'urticaire chronique et la rhinite allergique chronique.
Mode d’administration
Le comprimé pelliculé doit être pris par voie orale, sans être croqué, avec un peu de liquide au cours ou en dehors des repas. Il est recommandé de prendre la posologie quotidienne en une seule prise.
Hypersensibilité à la lévocétirizine, à d'autres dérivés de la pipérazine, ou à l'un des excipients mentionnés à la rubrique 6.1.
Insuffisance rénale sévère avec clairance de la créatinine inférieure à 10 ml/min.
4.4. Mises en garde spéciales et précautions d'emploi
L'utilisation du comprimé pelliculé est déconseillée chez l'enfant de moins de 6 ans car cette formulation ne permet pas d’adaptation posologique pour cette tranche d'âge. Il est recommandé d'utiliser une formulation pédiatrique de lévocétirizine.
Il est recommandé d’être prudent en cas d’ingestion d'alcool (voir Interactions).
Il convient d’être prudent chez les patients qui présentent des facteurs de prédisposition à une rétention urinaire (par ex. lésion médullaire, hyperplasie prostatique) parce que la lévocétirizine peut augmenter le risque de rétention urinaire.
Ce médicament contient du lactose. Son utilisation est déconseillée chez les patients présentant une intolérance au galactose, un déficit en lactase de Lapp ou un syndrome de malabsorption du glucose ou du galactose (maladies héréditaires rares).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les études réalisées avec la cétirizine (forme racémique) n'ont révélé aucune interaction cliniquement pertinente (avec la pseudoéphédrine, la cimétidine, le kétoconazole, l'érythromycine, l'azithromycine, le glipizide et le diazépam). De ce fait, aucune étude d'interaction n'a été réalisée avec la lévocétirizine (en particulier, pas d'étude avec les inducteurs du CYP3A4).
Il a été observé une légère diminution de la clairance de la cétirizine (16 %) avec la théophylline en prises répétées (400 mg par jour en une prise), tandis que la biodisponibilité de la théophylline n'est pas modifiée par une administration concomitante de cétirizine.
Dans une étude d'administration répétée de ritonavir (600 mg deux fois par jour) et de cétirizine (10 mg par jour), l'exposition à la cétirizine était augmentée d'environ 40 %, et la biodisponibilité du ritonavir était légèrement affectée (-11 %) par l'administration concomitante de cétirizine.
Le taux d'absorption de la lévocétirizine n'est pas diminué par l'alimentation, bien que sa vitesse d'absorption soit réduite.
Chez les patients sensibles, la prise concomitante d'alcool ou d'autres dépresseurs du système nerveux central avec la cétirizine ou la lévocétirizine pourrait avoir des effets sur le système nerveux central, même s’il a été démontré que la cétirizine racémique ne potentialise pas les effets de l'alcool.
4.6. Fertilité, grossesse et allaitement
Grossesse
Aucune donnée clinique concernant les grossesses exposées n’est disponible pour la lévocétirizine.
Les études sur l'animal n'ont pas montré d'effets nocifs directs ou indirects en termes de reprotoxicité (voir rubrique 5.3).
Par mesure de précaution, il est préférable d'éviter l'utilisation de lévocétirizine pendant la grossesse.
Allaitement
On ne sait pas si la lévocétirizine est excrétée dans le lait maternel. Un risque pour les nouveau-nés/nourissons ne peut être exclu.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Des études cliniques comparatives réalisées avec la lévocétirizine à la posologie recommandée n'ont pas révélé d'altération de la vigilance, de la réactivité, ou de l’aptitude à conduire des véhicules.
Cependant somnolence, fatigue et asthénie ont été décrits chez certains patients traités par lévocétirizine. Par conséquent, les patients susceptibles de conduire un véhicule, ou de manipuler un outil ou une machine potentiellement dangereux, devront évaluer au préalable leur réponse au traitement.
Au cours d'études cliniques menées chez des femmes et des hommes âgés de 12 à 71 ans, 15,1 % des patients du groupe lévocétirizine (5 mg) et 11,3 % des patients du groupe placebo ont présenté au moins un effet indésirable. 91,6 % de ces effets indésirables étaient d'intensité légère à modérée.
Dans les essais cliniques, le taux de sortie d'essai en raison d'évènements indésirables a été de 1,0 % (9/935 patients) dans le groupe lévocétirizine (5 mg), et de 1,8 % (14/771) dans le groupe placebo.
Au cours des essais cliniques, la lévocétirizine a été administrée à 935 sujets, à la posologie recommandée de 5 mg par jour.
Au cours de ces essais, l’incidence d’effets indésirables enregistrés à des fréquences de 1% ou plus (fréquent : ≥1/100, <1/10) sous lévocétirizine 5 mg ou sous placebo est indiquée dans le tableau ci-dessous:
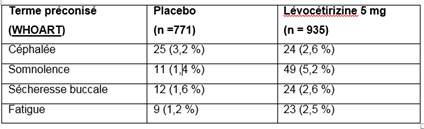
D’autres effets indésirables tels qu’asthénie et douleur abdominale ont été rarement observés (compris entre 1/1 000 à <1/100).
Un effet sédatif se manifestant notamment par une somnolence, une fatigue ou une asthénie a été observé dans l'ensemble plus fréquemment sous lévocétirizine 5 mg (8,1 %) qu'avec le placebo (3,1 %).
Des études en double aveugle et contrôlées par placebo ont été effectuées sur 243 enfants de 6–12 ans exposés à 5 mg de lévocétirizine par jour pendant diverses durées, de 1 à 13 semaines. Les effets indésirables suivants ont été rapportés à une fréquence supérieure ou égale à 1 % dans les groupes lévocétirizine 5 mg ou placebo :
|
Terme préféré |
Placebo (n=240) |
Lévocétirizine 5 mg (n=243) |
|
Céphalée |
5 (2,1 %) |
2 (0,8 %) |
|
Somnolence |
1 (0,4 %) |
7 (2,9 %) |
Données de pharmacovigilance
Les effets indésirables selon les données de pharmacovigilance sont classés par système d'organes et par fréquence. Les fréquences sont définies comme suit : très fréquent (≥1/10) ; fréquent (≥1/100 à <1/10) ; peu fréquent (≥1/1000 à <1/100) ; rare (≥1/10 000 à <1/1000) ; très rare (<1/10 000) ; fréquence indéterminée (ne peut être estimée à partir des données disponibles).
· Affections du système immunitaire :
Fréquence indéterminée : hypersensibilité, notamment anaphylaxie
· Troubles du métabolisme et de la nutrition :
Fréquence indéterminée : augmentation de l'appétit
· Affections psychiatriques :
Fréquence indéterminée : agression, agitation, hallucinations, dépression, insomnie, idées suicidaires
· Affections du système nerveux :
Fréquence indéterminée : convulsion, paresthésie, vertiges, syncope, tremblement, dysgueusie
· Affections de l'oreille et du labyrinthe :
Fréquence indéterminée : vertige
· Affections oculaires :
Fréquence indéterminée : troubles de la vision, vision brouillée, crises oculogyres.
· Affections cardiaques :
Fréquence indéterminée : palpitations, tachycardie
· Affections respiratoires, thoraciques et médiastinales :
Fréquence indéterminée : dyspnée
· Affections gastro-intestinales :
Fréquence indéterminée : nausée, vomissement
· Affections hépatobiliaires :
Fréquence indéterminée : hépatite
· Affections du rein et des voies urinaires :
Fréquence indéterminée : dysurie, rétention urinaire
· Affections de la peau et du tissu sous-cutané :
Fréquence indéterminée : angio-œdème, érythème pigmenté fixe, prurit, rash, urticaire
· Affections musculo-squelettiques et systémiques :
Fréquence indéterminée : myalgie
· Troubles généraux et anomalies au site d'administration :
Fréquence indéterminée : œdème
· Investigations :
Fréquence indéterminée : augmentation du poids, anomalies des tests de la fonction hépatique
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Symptômes
Les symptômes de surdosage peuvent inclure chez l’adulte, une somnolence et chez l'enfant, un état d'agitation suivi d’une somnolence.
Traitement du surdosage
Il n'existe pas d'antidote connu à la lévocétirizine.
En cas de surdosage, un traitement symptomatique ou de soutien sera entrepris en milieu spécialisé. Un lavage gastrique sera envisagé en cas d'ingestion récente. La lévocétirizine n'est pas éliminée par hémodialyse.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antihistaminique à usage systémique, dérivé de la pipérazine.
Code ATC : R06A E09.
La lévocétirizine, le R-énantiomère de la cétirizine, est un antagoniste puissant et sélectif des récepteurs périphériques H1.
Des études de liaison aux récepteurs ont révélé que la lévocétirizine a une forte affinité pour les récepteurs humains H1 (Ki = 3,2 nmol/l). La lévocétirizine a une affinité 2 fois supérieure à celle de la cétirizine (Ki = 6,3 nmol/l). La demi-vie de dissociation de la lévocétirizine des récepteurs H1 est de 115 ± 38 min. Il a été démontré que le taux d'occupation des récepteurs après administration unique de lévocétirizine est de 90 % après 4 h et de 57 % à 24 h.
Les études de pharmacodynamie menées chez le volontaire sain ont montré une activité comparable entre la cétirizine et la lévocétirizine administrée à demi dose, tant au niveau de la peau que du nez.
Les propriétés pharmacodynamiques de la lévocétirizine ont été étudiées dans des essais randomisés contrôlés.
Une étude a comparé les effets de la lévocétirizine 5 mg, la desloratadine 5 mg et un placebo, sur la réaction érythémato-papuleuse induite à l'histamine. Le traitement par lévocétirizine a significativement réduit papules et érythème (p < 0,001) avec une intensité maximale dans les 12 premières heures et maintenue pendant 24 h ce comparativement à la desloratadine et au placebo.
Dans une étude contrôlée contre placebo en chambre d'exposition pollinique, le délai d'action sur les symptômes a été de 1 heure après administration de 5 mg de lévocétirizine.
Les études menées in vitro (chambre de Boyden et technique sur culture cellulaire) montrent que la lévocétirizine inhibe in vitro la migration transendothéliale eotaxin-induite des éosinophiles à travers des cellules dermiques et bronchiques. Au cours d'une étude de pharmacodynamie expérimentale menée in vivo chez 14 patients (technique de chambre cutanée) trois effets inhibiteurs principaux ont été mis en évidence dans les premières 6 heures de la réaction induite par une exposition aux pollens: inhibition de la libération de VCAM-1, modulation de la perméabilité vasculaire et diminution du recrutement en éosinophiles.
L'efficacité et la sécurité de la lévocétirizine ont été démontrées au cours de plusieurs études cliniques en double aveugle, contrôlées, versus placebo, chez des patients présentant une rhinite allergique saisonnière ou perannuelle, ou persistante. La lévocétirizine a significativement amélioré les symptômes de la rhinite allergique, y compris l'obstruction nasale.
Une étude clinique réalisée sur 6 mois chez 551 patients adultes (dont 276 patients traités par lévocétirizine) présentant une rhinite allergique persistante (symptômes présents 4 jours par semaine pendant au moins 4 semaines consécutives) et sensibilisés aux acariens et aux pollens de graminées a démontré que la lévocétirizine 5 mg était significativement plus efficace que le placebo sur l'amélioration du score symptomatique global de la rhinite allergique sur toute la durée de l'étude, sans tachyphylaxie. Pendant toute la durée de l'étude, la lévocétirizine a significativement amélioré la qualité de vie des patients.
L'efficacité et la sécurité de la lévocétirizine sous forme comprimé ont été démontrées au cours de deux études cliniques contrôlées contre placebo chez des enfants de 6 à 12 ans présentant une rhinite allergique soit saisonnière soit perannuelle. Dans les deux études, le traitement par lévocétirizine a significativement amélioré les symptômes et la qualité de vie liée à l'état de santé.
La sécurité clinique chez l'enfant de moins de 6 ans a été établie par plusieurs études thérapeutiques de courte et de longue durée :
· une étude clinique dans laquelle 29 enfants de 2 à 6 ans avec rhinite allergique étaient traités avec 1,25 mg de lévocétirizine deux fois par jour pendant 4 semaines,
· une étude clinique dans laquelle 114 enfants de 1 à 5 ans avec rhinite allergique ou urticaire chronique idiopathique étaient traités avec 1,25 mg de lévocétirizine deux fois par jour pendant 2 semaines,
· une étude clinique dans laquelle 45 enfants de 6 à 11 mois avec rhinite allergique ou urticaire chronique idiopathique étaient traités avec 1,25 mg de lévocétirizine une fois par jour pendant 2 semaines,
· une étude clinique de longue durée (18 mois) avec administration de lévocétirizine à 255 sujets atopiques âgés de 12 à 24 mois au moment de l'inclusion.
Le profil de sécurité était similaire à celui observé dans les études de courte durée menées sur des enfants de 1 à 5 ans.
Dans une étude clinique contrôlée contre placebo réalisée chez 166 patients présentant une urticaire chronique idiopathique, 85 patients ont été traités par placebo et 5 mg de lévocétirizine ont été administrés à 81 patients une fois par jour pendant 6 semaines. Le traitement par lévocétirizine a significativement diminué la sévérité du prurit au cours de la première semaine et pendant toute la durée du traitement comparativement au placebo. La mesure de la qualité de vie sur l'échelle, Dermatology quality of life Index, a montré un effet significativement supérieur de la lévocétirizine 5 mg par rapport au placebo.
Les études cliniques visant à établir l'effet dans le traitement des manifestations allergiques cutanées ont été conduites chez des sujets atteints d'urticaire idiopathique chronique. La libération d'histamine étant le facteur déclenchant inducteur de manifestations cutanées urticariennes, l'efficacité en traitement symptomatique de la lévocétirizine peut être extrapolée aux autres formes d'urticaire.
Relations pharmacocinétique/pharmacodynamie :
L'effet sur les réactions cutanées provoquées par l'histamine n'est pas corrélé avec l'évolution des concentrations plasmatiques.
Les ECG n'ont pas montré d'effets de la lévocétirizine sur l'intervalle QT.
5.2. Propriétés pharmacocinétiques
La pharmacocinétique de la lévocétirizine est linéaire et indépendante du temps et de la dose avec une faible variabilité inter-individuelle. Les profils pharmacocinétiques de la lévocétirizine et de la cétirizine sont identiques. Aucune conversion chirale n'intervient au cours des processus d'absorption et d'élimination.
Absorption
La lévocétirizine est rapidement et largement absorbée après ingestion orale. Chez l'adulte, les concentrations plasmatiques maximales sont atteintes 0,9 h après la prise. L'état d'équilibre est atteint après 2 jours. Les concentrations plasmatiques maximales sont de 270 ng/ml et 308 ng/ml après administration, respectivement, d'une dose unique de 5 mg et de doses répétées de 5 mg par jour. La biodisponibilité est indépendante de la dose et n'est pas modifiée par la prise alimentaire, cependant, le pic de concentration est diminué et retardé.
Distribution
Aucune donnée n'est disponible chez l'homme concernant la diffusion tissulaire ou le passage de la barrière hémato-encéphalique de la lévocétirizine. Chez le rat et le chien, les plus fortes concentrations tissulaires ont été retrouvées au niveau du foie et des reins, les plus faibles au niveau du système nerveux central.
La fraction de lévocétirizine liée aux protéines plasmatiques est de 90 %. La distribution de la lévocétirizine est restreinte, puisque son volume de distribution est de 0,4 l/kg.
Biotransformation
Chez l'homme, la fraction de lévocétirizine métabolisée est inférieure à 14 % de la dose absorbée. Par conséquent, les différences résultant d'un polymorphisme génétique ou de la prise concomitante d'inhibiteurs enzymatiques sont considérés comme négligeables. Les voies métaboliques comprennent l'oxydation aromatique, la N et O - déalkylation et la conjugaison taurine. Les voies de déalkylation impliquent en premier lieu le CYP 3A4, l'oxydation implique des isoformes multiples et/ou non identifiées des CYP. La lévocétirizine n'a pas d'effet sur l'activité des isoenzymes CYP 1A2, 2C9, 2C19, 2D6, 2E1 et 3A4 à des concentrations nettement supérieures à celles atteintes après l'administration orale d'une dose de 5 mg.
En raison de sa faible métabolisation et de l'absence de potentiel inhibiteur du métabolisme, l'interaction de la lévocétirizine avec d'autres substances, ou vice-versa, est peu probable.
Élimination
Chez l'adulte, la demi-vie plasmatique est de 7,9 ± 1,9 heures. La clairance corporelle totale apparente moyenne est de 0,63 ml/min/kg. La principale voie d'élimination de la lévocétirizine et de ses métabolites est urinaire, représentant en moyenne 85,4 % de la dose absorbée. L'élimination par voie fécale ne représente que 12,9 % de la dose. La lévocétirizine est excrétée à la fois par filtration glomérulaire et par sécrétion tubulaire active.
Insuffisance rénale
La clairance corporelle apparente de la lévocétirizine est corrélée à la clairance de la créatinine. Il est par conséquent recommandé d'ajuster la fréquence d'administration de la lévocétirizine en fonction de la clairance de la créatinine, chez les patients atteints d'insuffisance rénale modérée à sévère. Chez les patients anuriques atteints d'insuffisance rénale au stade terminal, la clairance corporelle totale est réduite d'environ 80 % par rapport à celle d'un sujet normal. La quantité de lévocétirizine éliminée au cours d'une séance classique d'hémodialyse de 4 heures est inférieure à 10 %.
Population pédiatrique
Les données d'une étude pharmacocinétique pédiatrique avec administration orale d'une dose unique de 5 mg lévocétirizine à 14 enfants âgés de 6 à 11 ans et d'un poids corporel de 20 à 40 kg montrent que les valeurs de la Cmax et de l'ASC sont environ 2 fois plus élevées que celles observées chez les sujets adultes sains dans une étude croisée comparative. La Cmax moyenne était de 450 ng/ml et apparait après un temps moyen de 1,2 heure ; la clairance corporelle totale pondérée pour le poids est 30 % plus grande, et la demi-vie d'élimination est 24 % plus courte dans cette population pédiatrique que chez l'adulte. On ne dispose pas d'études pharmacocinétiques dédiées menées sur des patients pédiatriques de moins de 6 ans. Une analyse pharmacocinétique rétrospective de population a porté sur 324 sujets (181 enfants de 1 à 5 ans, 18 enfants de 6 à 11 ans et 124 adultes de 18 à 55 ans) qui avaient reçu une dose unique ou des doses répétées de 1,25 à 3 mg de lévocétirizine. Les données issues de cette analyse ont montré que l'administration de 1,25 mg une fois par jour aux enfants de 6 mois à 5 ans induirait des concentrations plasmatiques similaires à celles des adultes traités avec 5 mg une fois par jour.
Patients âgés
Les données pharmacocinétiques pour les sujets âgés sont limitées. Après administration orale répétée de 30 mg de lévocétirizine une fois par jour pendant 6 jours chez 9 sujets âgés (65–74 ans), la clairance corporelle totale était environ 33 % inférieure à celle des jeunes adultes. La disponibilité du racémique, la cétirizine, dépend de la fonction rénale plutôt que de l'âge.
Ce résultat s’applique également à la lévocétirizine puisque la lévocétirizine et la cétirizine sont toutes deux principalement éliminées par voie urinaire. La posologie de lévocétirizine doit donc être ajustée chez les patients âgés selon leur fonction rénale.
Genre
Les résultats pharmacocinétiques de 77 patients (40 hommes, 37 femmes) ont été étudiés pour évaluer l’influence du genre. La demi-vie était légèrement plus courte chez la femme (7,08 ± 1,72 h) que chez l'homme (8,62 ± 1,84 h) ; après ajustement pour le poids corporel, la clairance après administration orale à la femme (0,67 ± 0,16 ml/min/kg) est cependant similaire à celle observée chez l'homme (0,59 ± 0,12 ml/min/kg).
Les mêmes posologies et mode d'administration sont applicables aux hommes et aux femmes dont la fonction rénale est normale.
Type ethnique
L'effet du type ethnique sur la pharmacocinétique de la lévocétirizine n'a pas été étudié. La lévocétirizine est éliminée principalement par excrétion rénale et il n'existe pas de différences ethniques importantes quant à la clairance de la créatinine ; les paramètres pharmacocinétiques ne devraient pas différer en fonction du type ethnique. On n'a pas observé de différences ethniques des paramètres pharmacocinétiques pour le racémique, la cétirizine.
Insuffisance hépatique
Les paramètres pharmacocinétiques de la lévocétirizine chez les patients avec insuffisance hépatique n'ont pas été étudiés. Les patients présentant une maladie hépatique chronique (hépatocellulaire, cholestatique, cirrhose biliaire) recevant une dose unique de 10 ou 20 mg du racémique, la cétirizine, présentent une augmentation de 50 % de la demi-vie et une diminution de 40 % de la clairance, par rapport aux sujets sains.
5.3. Données de sécurité préclinique
Les données précliniques, issues d'études classiques de sécurité pharmacologique, de toxicité chronique, de génotoxicité, de potentiel carcinogène et de toxicité sur la reproduction, n'ont pas révélé de risque potentiel particulier pour l'être humain.
Noyau du comprimé : cellulose microcristalline, lactose monohydraté, silice colloïdale anhydre, stéarate de magnésium.
Pelliculage : hypromellose, dioxyde de titane (E171), talc, macrogol 400.
Sans objet.
3 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
14, 20, 28, 40 ou 60 comprimés sous plaquette (PVDC/PVC – Aluminium).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières pour l’élimination.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
RANBAXY PHARMACIE GENERIQUES
11/15 QUAI DE DION BOUTON
92800 PUTEAUX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 300 113 8 6 : 14 comprimés sous plaquettes (PVC/PVDC/Aluminium)
· 34009 275 959 7 4 : 20 comprimés sous plaquettes (PVC/PVDC/Aluminium)
· 34009 300 114 0 9 : 28 comprimés sous plaquettes (PVC/PVDC/Aluminium)
· 34009 275 960 5 6 : 40 comprimés sous plaquettes (PVC/PVDC/Aluminium)
· 34009 275 961 1 7 : 60 comprimés sous plaquettes (PVC/PVDC/Aluminium)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste II.
|
|
| Plan du site | Accessibilité | Contact | Téléchargement | Declaration de confidentialité | Service-Public.fr | Legifrance | Gouvernement.fr |