Base de données publique
des médicaments
Visiter [medicaments.gouv.fr] ![Visiter [medicaments.gouv.fr]](/img/icone_lien.png "Visiter [medicaments.gouv.fr] - nouvelle fenêtre")



SPIRIVA RESPIMAT 2,5 microgrammes/dose, solution à inhaler - Résumé des caractéristiques du produit |
 |
|
ANSM - Mis à jour le : 01/02/2023
SPIRIVA RESPIMAT 2,5 microgrammes/dose, solution à inhaler
2. COMPOSITION QUALITATIVE ET QUANTITATIVE 
Tiotropium…………………………………………………………………….………………2,5 microgrammes
Sous forme de bromure de tiotropium monohydraté…………………………….…...3,124 microgrammes
pour une bouffée délivrée à la sortie de l’embout buccal
La dose délivrée par bouffée correspond à la quantité de tiotropium délivré à la sortie de l’embout buccal.
La dose journalière correspond à 2 bouffées.
Excipient à effet notoire : ce médicament contient 0,0011 mg de chlorure de benzalkonium par bouffée.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution à inhaler limpide et incolore.
4.1. Indications thérapeutiques
Bronchopneumopathie chronique obstructive (BPCO)
SPIRIVA RESPIMAT est indiqué en traitement bronchodilatateur continu destiné à soulager les symptômes des patients présentant une bronchopneumopathie chronique obstructive.
Asthme
SPIRIVA RESPIMAT est indiqué en traitement bronchodilatateur additionnel continu chez des patients de 6 ans et plus atteints d’asthme sévère qui ont présenté au cours de l’année précédente une ou plusieurs exacerbations sévères d’asthme (voir rubriques 4.2 et 5.1).
4.2. Posologie et mode d'administration
Voie inhalée exclusivement.
La cartouche ne peut être insérée et utilisée qu’avec l’inhalateur RESPIMAT.
La dose journalière est de 2 bouffées successives (soit 2 pressions sur le bouton) administrées à l’aide de l’inhalateur RESPIMAT.
La posologie recommandée chez l’adulte est de 5 microgrammes de tiotropium, soit 2 bouffées successives (soit 2 pressions sur le bouton) administrées à l’aide de l’inhalateur RESPIMAT, en une prise par jour, à heure fixe dans la journée.
Ne pas dépasser la dose recommandée.
Dans le traitement de l’asthme, le bénéfice complet peut n’apparaître qu’après plusieurs jours de traitement. Chez les patients adultes atteints d’asthme sévère, le tiotropium doit être utilisé en association à un corticostéroïde inhalé (CSI) (≥ 800 µg de budésonide/jour ou équivalent) et au moins un traitement de contrôle.
Populations particulières
Sujets âgés : le bromure de tiotropium peut être utilisé chez les sujets âgés sans adaptation de la posologie.
Insuffisance rénale : en cas d’insuffisance rénale, le bromure de tiotropium peut être utilisé sans adaptation de la posologie. En cas d’insuffisance rénale modérée à sévère (clairance de la créatinine £ 50 ml/min) voir rubriques 4.4 et 5.2.
Insuffisance hépatique : en cas d’insuffisance hépatique, le bromure de tiotropium peut être utilisé sans adaptation de la posologie (voir rubrique 5.2).
Population pédiatrique
Asthme : la posologie recommandée chez les patients âgés de 6 à 17 ans est de 5 microgrammes de tiotropium, soit 2 bouffées consécutives administrées à l’aide de l’inhalateur RESPIMAT, en une prise par jour, à heure fixe dans la journée.
Chez les adolescents (12-17 ans) atteints d’asthme sévère, le tiotropium doit être utilisé en association à un CSI (> 800-1600 µg de budésonide/jour ou équivalent) et un traitement de contrôle ou en association à un CSI (400-800 µg de budésonide/jour ou équivalent) et deux traitements de contrôle.
Chez les enfants (6-11 ans) atteints d’asthme sévère, le tiotropium doit être utilisé en association à un CSI (> 400 µg de budésonide/jour ou équivalent) et un traitement de contrôle ou en association à un CSI (200‑400 µg de budésonide/jour ou équivalent) et deux traitements de contrôle.
La sécurité et l’efficacité de SPIRIVA RESPIMAT chez les enfants âgés de 6 à 17 ans atteints d’asthme modéré n’ont pas encore été établies. La sécurité et l’efficacité de SPIRIVA RESPIMAT chez les enfants de moins de 6 ans n’ont pas été établies. Les données actuellement disponibles sont décrites dans les rubriques 5.1 et 5.2 mais aucune recommandation sur la posologie ne peut être faite.
BPCO : il n’y a pas de justification à l’utilisation de SPIRIVA RESPIMAT chez les enfants et les adolescents de moins de 18 ans.
Mucoviscidose : la sécurité et l’efficacité de SPIRIVA RESPIMAT n’ont pas été établies (voir rubriques 4.4 et 5.1).
Mode d’administration
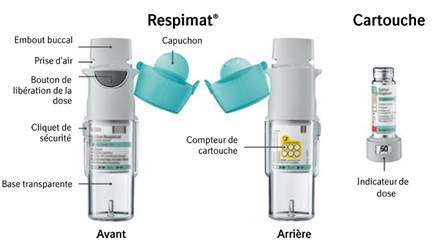
Les patients doivent lire les instructions d’utilisation de l’inhalateur RESPIMAT réutilisable avant de commencer à utiliser SPIRIVA RESPIMAT.
Pour une administration correcte du médicament, le médecin ou un autre professionnel de santé devra s’assurer du bon usage de l’appareil par le patient.
Mode d’emploi pour la manipulation et l’utilisation de l’inhalateur RESPIMAT réutilisable
Les enfants doivent utiliser SPIRIVA RESPIMAT avec l’aide d’un adulte.
Le patient utilisera cet inhalateur UNE SEULE FOIS PAR JOUR. A chaque utilisation, prendre 2 BOUFFEES (soit 2 pressions sur le bouton de libération de la dose).
· Si SPIRIVA RESPIMAT n’a pas été utilisé pendant plus de 7 jours, exercer une pression sur le bouton pour libérer une bouffée vers le sol.
· Si SPIRIVA RESPIMAT n’a pas été utilisé pendant plus de 21 jours, répéter les étapes 4 à 6 « Préparation pour l’utilisation » ci-dessous jusqu'à ce qu'un nuage apparaisse. Puis, répéter les étapes 4 à 6 trois fois supplémentaires.
Comment entretenir l'inhalateur RESPIMAT réutilisable
Nettoyer l’embout buccal, y compris la partie métallique à l’intérieur de l’embout buccal, en utilisant uniquement un linge ou un tissu humide, au moins une fois par semaine.
L’apparition d’une décoloration mineure de l’embout buccal n’affecte pas les performances de l'inhalateur RESPIMAT réutilisable.
Si nécessaire, essuyer l’extérieur de l'inhalateur RESPIMAT réutilisable avec un linge humide.
Quand se procurer un nouvel inhalateur
Une fois que le patient a utilisé l’inhalateur avec 6 cartouches, il doit se procurer une nouvelle boîte de SPIRIVA RESPIMAT contenant un inhalateur.


Préparation pour l’utilisation
|
1. Retirer la base transparente · Maintenir le capuchon fermé. · Appuyer sur le cliquet de sécurité tout en retirant la base transparente avec l'autre main.
|
|
|
2. Insérer la cartouche · Insérer la cartouche dans l’inhalateur. · Placer l’inhalateur sur une surface solide et pousser fermement jusqu’à ce qu’il se mette en place. |
|
|
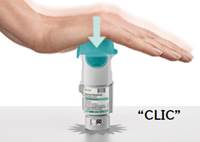
3. Surveiller le nombre de cartouches utilisées et remettre la base transparente · Cocher la case sur l’étiquette de l’inhalateur pour comptabiliser le nombre de cartouches utilisées. · Remettre en place la base transparente jusqu’à entendre un déclic.
|
|
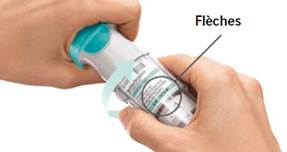
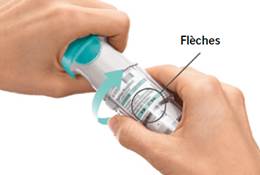
4. Tourner · Maintenir le capuchon fermé. · Tourner la base transparente dans la direction des flèches imprimées sur l’étiquette jusqu’à entendre un déclic (un demi-tour).
|
|
|
5. Ouvrir · Ouvrir complètement le capuchon. |
|
|
6. Presser · Diriger l’inhalateur en direction du sol. · Presser le bouton de libération de la dose. · Fermer le capuchon. · Répéter les étapes 4 à 6 jusqu’à ce qu’un nuage apparaisse. · A l’apparition du nuage, répéter les étapes 4 à 6 trois fois supplémentaires. L’inhalateur est maintenant prêt à être utilisé et 60 bouffées pourront être libérées (soit 30 doses). |
|
Utilisation quotidienne
|
TOURNER · Maintenir le capuchon fermé. · TOURNER la base transparente dans la direction des flèches imprimées sur l’étiquette jusqu’à entendre un déclic (un demi-tour).
|
|
|
OUVRIR · OUVRIR complétement le capuchon |
|
|
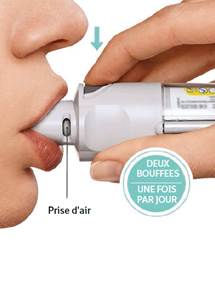
PRESSER · Expirer lentement et complètement en dehors du dispositif. · Fermer les lèvres autour de l’embout buccal sans recouvrir les prises d’air. Diriger l'embout buccal de l’inhalateur vers le fond de la gorge. · Tout en inspirant lentement et profondément par la bouche, PRESSER le bouton de libération de la dose et continuer d’inspirer lentement aussi longtemps que possible sans être gêné. · Bloquer la respiration pendant 10 secondes ou aussi longtemps que possible sans être gêné. · Répéter TOURNER, OUVRIR, PRESSER sur le bouton pour délivrer la 2ème bouffée (2 bouffées successives au total). · Fermer le capuchon jusqu’à la prochaine utilisation de l'inhalateur.
|
|
Quand remplacer la cartouche de SPIRIVA RESPIMAT
L’indicateur de dose indique le nombre de bouffées restantes dans la cartouche.
![]()
60 bouffées (60 pressions) restantes.
![]()
Moins de 10 bouffées (10 pressions) restantes. Se procurer une nouvelle cartouche.
![]() La cartouche est vide. Tourner la base transparente afin de la retirer. L’inhalateur est maintenant en position verrouillée. Retirer la cartouche de l’inhalateur. Insérer une nouvelle cartouche jusqu’à entendre un déclic (se référer à l’étape 2). La nouvelle cartouche dépassera davantage que la toute première cartouche (continuer avec l'étape 3). Ne pas oublier de remettre la base transparente pour déverrouiller l'inhalateur.
La cartouche est vide. Tourner la base transparente afin de la retirer. L’inhalateur est maintenant en position verrouillée. Retirer la cartouche de l’inhalateur. Insérer une nouvelle cartouche jusqu’à entendre un déclic (se référer à l’étape 2). La nouvelle cartouche dépassera davantage que la toute première cartouche (continuer avec l'étape 3). Ne pas oublier de remettre la base transparente pour déverrouiller l'inhalateur.
4.4. Mises en garde spéciales et précautions d'emploi
Le chlorure de benzalkonium contenu dans ce médicament peut provoquer une respiration sifflante et des difficultés respiratoires. Les patients asthmatiques ont un risque élevé de présenter ces événements indésirables.
Le bromure de tiotropium est un traitement bronchodilatateur continu de longue durée d’action en une prise par jour. Il ne doit pas être utilisé comme un médicament de première intention pour le traitement des épisodes aigus de bronchospasme ou pour le soulagement des symptômes aigus. En cas de crise d’asthme, un bêta-2-agoniste d’action rapide doit être utilisé.
SPIRIVA RESPIMAT ne doit pas être utilisé en monothérapie dans le traitement de l’asthme. Lors de la mise en route du traitement par SPIRIVA RESPIMAT, les patients asthmatiques doivent être informés qu’ils doivent continuer leur traitement anti-inflammatoire par corticostéroïdes inhalés sans le modifier même si les symptômes s’améliorent.
Des réactions d’hypersensibilité immédiate peuvent survenir après l’inhalation du bromure de tiotropium.
En raison de son activité anticholinergique, le bromure de tiotropium doit être utilisé avec prudence en cas de glaucome à angle fermé, d'hypertrophie de la prostate ou de rétrécissement du col de la vessie.
D’une façon générale, l’administration par voie inhalée des médicaments peut déclencher un bronchospasme.
Le tiotropium doit être utilisé avec prudence chez les patients ayant eu un infarctus du myocarde au cours des six derniers mois, une arythmie cardiaque instable ou menaçant le pronostic vital, une arythmie cardiaque ayant nécessité une intervention ou une modification du traitement médicamenteux au cours de l’année précédente, ou chez les patients ayant été hospitalisés pour une insuffisance cardiaque (NYHA classe III ou IV) au cours de l’année précédente. Les patients ayant de tels antécédents ont été exclus des essais cliniques et un retentissement lié à l'activité anticholinergique du tiotropium dans ces situations n'est pas exclu.
Les concentrations plasmatiques de bromure de tiotropium augmentent chez les patients présentant une fonction rénale altérée. Par conséquent, en cas d'insuffisance rénale modérée à sévère (clairance de la créatinine £ 50 ml/min), le bromure de tiotropium ne doit être utilisé que si le bénéfice attendu est supérieur au risque potentiel. A ce jour, il n'y a pas d'expérience à long terme avec le tiotropium chez les patients présentant une insuffisance rénale sévère (voir rubrique 5.2).
Il conviendra d’avertir les patients du risque de déclenchement ou d’aggravation d'un glaucome à angle fermé, de douleur ou gêne oculaire, de vision trouble transitoire avec halo visuel ou images colorées associée à une rougeur oculaire et un œdème cornéo-conjonctival, en cas de projection intraoculaire du produit. Si un ou plusieurs de ces symptômes oculaires apparaissent, les patients doivent interrompre l’utilisation du bromure de tiotropium et consulter immédiatement un médecin.
La sécheresse buccale observée avec les traitements anticholinergiques en général, peut à long terme favoriser la survenue de caries dentaires.
La posologie du bromure de tiotropium ne doit pas dépasser une prise par jour (voir rubrique 4.9).
SPIRIVA RESPIMAT n’est pas recommandé dans le traitement de la mucoviscidose. En cas d’utilisation chez des patients atteints de mucoviscidose, SPIRIVA RESPIMAT peut augmenter les signes et symptômes de la mucoviscidose (par exemple : événements indésirables graves, exacerbations pulmonaires, infections des voies respiratoires).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L’utilisation des beta2-agonistes de longue durée d'action (LABA) ou des corticostéroïdes inhalés (CSI) n’a pas modifié de façon significative l’exposition au tiotropium.
L’administration concomitante de bromure de tiotropium avec d'autres médicaments à activité anticholinergique n'a pas été étudiée et n’est par conséquent, pas recommandée.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données disponibles sur l’utilisation du tiotropium chez la femme enceinte sont très limitées. Les études effectuées chez l’animal n’ont pas mis en évidence d’effets délétères directs ou indirects sur la reproduction à des doses cliniquement significatives (voir rubrique 5.3). Par mesure de précaution, il est préférable d’éviter l’utilisation de SPIRIVA RESPIMAT pendant la grossesse.
Allaitement
L’excrétion du bromure de tiotropium dans le lait maternel n’a pas été établie. Bien que l’excrétion du bromure de tiotropium n’ait été retrouvée qu’en faible quantité dans le lait maternel au cours des études réalisées chez les rongeurs, l’utilisation de SPIRIVA RESPIMAT n’est pas recommandée au cours de l’allaitement. Le bromure de tiotropium est un principe actif de longue durée d’action. La décision de poursuivre ou non l’allaitement ou bien d'interrompre le traitement par SPIRIVA RESPIMAT doit tenir compte du bénéfice de l’allaitement chez l’enfant et du bénéfice du traitement par SPIRIVA RESPIMAT chez la femme qui allaite.
Fertilité
Aucune donnée clinique sur la fertilité n’est disponible avec le tiotropium. Une étude non clinique effectuée chez l'animal avec du tiotropium n’a pas montré d’effets indésirables sur la fertilité (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Un grand nombre des effets indésirables rapportés peuvent être attribués aux propriétés anticholinergiques du bromure de tiotropium.
Tableau résumé des effets indésirables
Les fréquences de survenue des effets indésirables présentés ci-dessous ont été déterminées sur la base de l’incidence des effets indésirables observés chez les patients traités par le tiotropium (c’est-à-dire les événements imputables au tiotropium), établie à partir du regroupement des données de 7 études cliniques dans la BPCO (3282 patients) et de 12 études cliniques dans l’asthme chez les adultes et dans la population pédiatrique (1930 patients), contrôlées contre placebo, dont les durées de traitement étaient comprises entre 4 semaines et 1 an.
La fréquence est définie selon la classification conventionnelle :
Très fréquent (≥1/10) ; fréquent (≥1/100 à <1/10) ; peu fréquent (≥1/1000 à <1/100) ; rare (≥1/10 000 à <1/1000) ; très rare (<1/10 000) ; fréquence indéterminée (fréquence ne pouvant être estimée sur la base des données disponibles).
|
Classe d'organes/Terme MedDRA recommandé |
Fréquence dans la BPCO |
Fréquence dans l’asthme |
|
Troubles du métabolisme et de la nutrition |
|
|
|
Déshydratation |
Indéterminée |
Indéterminée |
|
Troubles du système nerveux |
|
|
|
Etourdissements |
Peu fréquent |
Peu fréquent |
|
Céphalées |
Peu fréquent |
Peu fréquent |
|
Insomnies |
Rare |
Peu fréquent |
|
Troubles oculaires |
|
|
|
Glaucome |
Rare |
Indéterminée |
|
Augmentation de la pression intraoculaire |
Rare |
Indéterminée |
|
Vision trouble |
Rare |
Indéterminée |
|
Troubles cardiaques |
|
|
|
Fibrillation auriculaire |
Rare |
Indéterminée |
|
Palpitations |
Rare |
Peu fréquent |
|
Tachycardie supraventriculaire |
Rare |
Indéterminée |
|
Tachycardie |
Rare |
Indéterminée |
|
Troubles respiratoires, thoraciques et médiastinaux |
|
|
|
Toux |
Peu fréquent |
Peu fréquent |
|
Pharyngite |
Peu fréquent |
Peu fréquent |
|
Dysphonie |
Peu fréquent |
Peu fréquent |
|
Epistaxis |
Rare |
Rare |
|
Bronchospasme |
Rare |
Peu fréquent |
|
Laryngite |
Rare |
Indéterminée |
|
Sinusite |
Indéterminée |
Indéterminée |
|
Troubles gastro-intestinaux |
|
|
|
Sécheresse buccale |
Fréquent |
Peu fréquent |
|
Constipation |
Peu fréquent |
Rare |
|
Candidose oropharyngée |
Peu fréquent |
Peu fréquent |
|
Dysphagie |
Rare |
Indéterminée |
|
Reflux gastro-œsophagien |
Rare |
Indéterminée |
|
Caries dentaires |
Rare |
Indéterminée |
|
Gingivite |
Rare |
Rare |
|
Glossite |
Rare |
Indéterminée |
|
Stomatite |
Indéterminée |
Rare |
|
Occlusion intestinale, y compris iléus paralytique |
Indéterminée |
Indéterminée |
|
Nausées |
Indéterminée |
Indéterminée |
|
Troubles cutanés et du tissu sous-cutané, Troubles du système immunitaire |
|
|
|
Eruption cutanée |
Peu fréquent |
Peu fréquent |
|
Prurit |
Peu fréquent |
Rare |
|
Œdème de Quincke |
Rare |
Rare |
|
Urticaire |
Rare |
Rare |
|
Infection cutanée/ulcération cutanée |
Rare |
Indéterminée |
|
Sécheresse cutanée |
Rare |
Indéterminée |
|
Hypersensibilité (y compris réactions immédiates) |
Indéterminée |
Rare |
|
Réaction anaphylactique |
Indéterminée |
Indéterminée |
|
Troubles musculosquelettiques et systémiques |
|
|
|
Gonflement articulaire |
Indéterminée |
Indéterminée |
|
Troubles rénaux et des voies urinaires |
|
|
|
Rétention d’urine |
Peu fréquent |
Indéterminée |
|
Dysurie |
Peu fréquent |
Indéterminée |
|
Infection urinaire |
Rare |
Rare |
Description de certains effets indésirables
Dans les essais cliniques contrôlés menés dans la BPCO, les effets indésirables le plus fréquemment observé ont été les effets indésirables de type anticholinergiques tels que la sécheresse buccale survenue chez environ 2,9% des patients. Dans ceux menés dans l’asthme, la fréquence de la sécheresse buccale a été de 0,83%.
Dans 7 essais cliniques menés dans la BPCO, la sécheresse buccale a été à l’origine de 3 arrêts de traitement parmi 3282 patients traités par le tiotropium (soit 0,1% des patients traités). Aucune interruption du traitement due à une sécheresse buccale n’a été rapportée dans les 12 essais cliniques menés dans l’asthme (1930 patients).
Les effets indésirables graves liés aux effets anticholinergiques incluent : glaucome, constipation, occlusion intestinale y compris iléus paralytique et rétention urinaire.
Population pédiatrique
La base de données de sécurité inclut 560 patients pédiatriques (296 patients âgés de 1 à 11 ans et 264 patients âgés de 12 à 17 ans) de 5 études cliniques contrôlées contre placebo ayant des périodes de traitement de 12 semaines à 1 an. La fréquence, le type et la sévérité des effets indésirables dans la population pédiatrique ont été similaires à ceux observés chez les adultes.
Autres populations particulières
Le risque de survenue des effets anticholinergiques peut augmenter avec l’âge.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
L'administration de doses élevées de bromure de tiotropium peut déclencher l'apparition de signes et symptômes de type anticholinergique.
Cependant, l'inhalation par des volontaires sains d'une dose unique allant jusqu'à 340 microgrammes de bromure de tiotropium n'a entraîné aucun effet indésirable systémique de type anticholinergique. En outre, aucun effet indésirable significatif, hormis une sécheresse buccale, pharyngée ou de la muqueuse nasale, n’a été observé après 14 jours d’administration de doses atteignant 40 microgrammes de tiotropium solution à inhaler chez des volontaires sains, à l’exception d’une diminution importante de la salivation à partir du 7ème jour.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Autres médicaments pour les syndromes obstructifs des voies aériennes par inhalation, anticholinergiques, code ATC : R03B B04.
Mécanisme d’action
Le bromure de tiotropium est un antagoniste spécifique de longue durée d’action des récepteurs muscariniques, qui montre une affinité similaire pour les sous-types de récepteurs muscariniques M1 à M5. Dans les voies aériennes, le bromure de tiotropium se fixe de façon compétitive et réversible sur les récepteurs M3 des muscles lisses bronchiques, et inhibe les effets cholinergiques (bronchoconstriction) de l’acétylcholine, entraînant ainsi une relaxation des muscles lisses bronchiques. L’effet est dose-dépendant et persiste plus de 24 heures. Anticholinergique de type ammonium quaternaire, le bromure de tiotropium exerce une action topique (bronchique) sélective lorsqu’il est administré par inhalation, et offre ainsi une marge thérapeutique acceptable avant l’apparition des effets anticholinergiques systémiques.
Effets pharmacodynamiques
La dissociation du tiotropium des récepteurs M3 notamment est très lente, lui conférant une demi-vie de dissociation significativement plus longue que l’ipratropium. Le tiotropium se dissocie plus rapidement des récepteurs M2 que des récepteurs M3, comme le suggèrent les études in vitro, marquant une sélectivité plus importante (exprimée de façon cinétique) pour les récepteurs de type M3 par rapport aux récepteurs de type M2. La forte activité, la très lente dissociation des récepteurs et la sélectivité topique de l’administration par inhalation se traduisent sur le plan clinique par une bronchodilatation significative et prolongée chez les patients atteints de BPCO et d’asthme.
Efficacité et sécurité clinique dans le traitement de la BPCO
Le programme de développement clinique de phase III de SPIRIVA RESPIMAT a été composé de plusieurs études randomisées et en double aveugle (2 études d’un an, 2 études de 12 semaines et 2 études de 4 semaines) portant sur 2 901 patients présentant une BPCO (1 038 recevant la dose de 5 µg de tiotropium). Le développement sur un an a été composé de deux essais contrôlés contre placebo. Les 2 études de 12 semaines étaient toutes 2 menées comparativement à un groupe contrôle placebo et un groupe de traitement actif (ipratropium). L’ensemble des 6 études a comporté des mesures de la fonction pulmonaire. De plus, les deux études d’un an ont inclus les mesures de la dyspnée, de la qualité de vie liée à l’état de santé et la survenue des exacerbations.
Etudes contrôlées contre placebo
Fonction pulmonaire
Le tiotropium solution à inhaler, administré une fois par jour, a apporté des améliorations significatives de la fonction pulmonaire (volume expiratoire maximum par seconde et capacité vitale forcée) dans un délai de 30 minutes suivant la première dose, par rapport au placebo (amélioration moyenne du VEMS à 30 minutes : 0,113 litres ; intervalle de confiance IC à 95% : 0,102 à 0,125 litres, p< 0,0001). A l’état d’équilibre, l’amélioration de la fonction pulmonaire a été maintenue pendant 24 heures, par rapport au placebo (amélioration moyenne du VEMS: 0,122 litres ; IC95% : 0,106 à 0,138 litres,
p< 0,0001). L’état d’équilibre pharmacodynamique a été atteint en une semaine.
SPIRIVA RESPIMAT a significativement amélioré, comparativement au placebo, le débit expiratoire de pointe (DEP) du matin et du soir, mesuré quotidiennement par les patients, (amélioration moyenne du DEP le matin : 22 l/min ; IC95% : 18 à 55 l/min, p< 0,0001 ; le soir 26 l/min ; IC95% : 23 à 30 l/min, p<0,0001). L’utilisation de SPIRIVA RESPIMAT a entraîné une réduction de l’utilisation du traitement bronchodilatateur de secours par rapport au placebo (réduction moyenne de 0,66 prise par jour, IC95% : 0,51 à 0,81 prise par jour, p < 0,0001).
L’effet bronchodilatateur de SPIRIVA RESPIMAT s’est maintenu pendant une période d’administration d’un an sans signe d’induction d’une tolérance.
Dyspnée, qualité de vie, exacerbations de BPCO au cours d’études d’une durée d’un an
Dyspnée
SPIRIVA RESPIMAT a amélioré de façon significative la dyspnée (évaluée par l’index de dyspnée de transition) comparativement au placebo (amélioration moyenne de 1,05 unités ; IC95% : 0,73 à 1,38 unités, p<0,0001). Cette amélioration s’est maintenue pendant toute la période de traitement.
Qualité de vie
L’amélioration du score total moyen de la qualité de vie évalué par les patients eux-mêmes au moyen du questionnaire respiratoire de St George (SGRQ) obtenue avec SPIRIVA RESPIMAT à la fin des deux études d’un an a été de 3,5 unités par rapport au placebo (IC95% : 2,1 à 4,9, p<0,0001). Une diminution de 4 unités est considérée comme cliniquement significative.
Exacerbations de BPCO
Dans trois études cliniques randomisées d’une durée d’un an, conduites en double aveugle, contre placebo, le traitement par SPIRIVA RESPIMAT a entraîné une réduction significative du risque d’exacerbations de BPCO par rapport au placebo. Les exacerbations de BPCO ont été définies de la façon suivante : « association d’au moins deux événements ou symptômes respiratoires persistants pendant 3 jours ou plus et ayant conduit à un changement de traitement (prescription d’antibiotiques et/ou de corticostéroïdes systémiques et/ou modification significative du traitement à visée respiratoire) ».
Le traitement par SPIRIVA RESPIMAT a entraîné une réduction du risque d’hospitalisations en relation avec une exacerbation de BPCO (statistiquement significatif dans l’étude sur les exacerbations dont la puissance a été considérée comme appropriée). L’analyse regroupant les données de deux études de phase III et l’analyse distincte d’une étude supplémentaire portant sur les exacerbations sont présentées dans le Tableau 1. Tous les traitements à visée respiratoire, à l’exception des anticholinergiques et des ß-mimétiques d’action prolongée, étaient autorisés en traitements concomitants, consistant en ß-mimétiques d’action rapide, corticostéroïdes inhalés et xanthines. Les ß-mimétiques d’action prolongée étaient autorisés en traitement complémentaire dans l’étude sur les exacerbations.
Tableau 1 : Analyse statistique des exacerbations de BPCO et des exacerbations de BPCO ayant nécessité une hospitalisation chez les patients présentant une BPCO modérée à très sévère :
|
Etude (NSpiriva, Nplacebo) |
Critère |
SPIRIVA |
Placebo |
% de réduction du risque (IC à 95 %)a |
Valeur de p |
|
Analyse combinée des études de phase III d'un an d |
Nombre de jours avant la première exacerbation de BPCO |
160a |
86a |
29 |
< 0,0001b |
|
(670, 653) |
Incidence moyenne d'exacerbations par patient-année |
0,78c |
1,00c |
22 |
0,002c |
|
|
Délai avant la première exacerbation de BPCO ayant nécessité une hospitalisation |
|
|
25 |
0,20b |
|
|
Incidence moyenne d'exacerbations ayant nécessité une hospitalisation par patient-année |
0,09c |
0,11c |
20 |
0,096c |
|
Etude sur les exacerbations de phase III b d'un an |
Nombre de jours avant la première exacerbation de BPCO |
169a |
119a |
31 |
< 0,0001b |
|
(1939, 1953) |
Incidence moyenne d'exacerbations par patient-année |
0,69c |
0,87c |
21 |
< 0,0001c |
|
|
Délai avant la première exacerbation de BPCO ayant nécessité une hospitalisation |
|
|
27 |
0,003b |
|
|
Incidence moyenne des exacerbations ayant nécessité une hospitalisation par patient-année |
0,12c |
0,15c |
19 (7 à 30)c |
0,004c |
a Délai avant le premier événement: nombre de jours sous traitement écoulés avant que 25 % des patients présentent au moins une exacerbation de BPCO /hospitalisation pour exacerbation de BPCO. Dans l'étude A, 25 % des patients sous placebo ont présenté une exacerbation au jour 112 alors que 25 % des patients traités par SPIRIVA RESPIMAT ont présenté une exacerbation au jour 173
(p = 0,09). Dans l'étude B, 25 % des patients sous placebo ont présenté une exacerbation au jour 74 alors que 25 % des patients traités par SPIRIVA RESPIMAT 2,5 microgrammes/dose, solution à inhaler ont présenté une exacerbation au jour 149 (p< 0, 0001).
b Les risques relatifs ont été estimés avec un modèle aléatoire proportionnel de Cox. La réduction du risque exprimé en pourcentage est calculée avec la formule suivante : 100 (1 - risque relatif).
c Régression de Poisson. La réduction du risque est calculée avec la formule suivante : 100 (1 - rapport des taux).
d Le regroupement pour l’analyse des données était prévu initialement. Les paramètres mesurant les exacerbations ont été significativement améliorés dans l’analyse séparée des 2 études d’un an.
Etude à long terme du tiotropium contrôlée par une substance active
Une étude à long terme menée à grande échelle, randomisée, en double aveugle, contrôlée, incluant une période d’observation de 3 ans, a comparé l’efficacité et la sécurité de SPIRIVA RESPIMAT et de SPIRIVA HANDIHALER (5711 patients recevant SPIRIVA RESPIMAT ; 5694 patients recevant SPIRIVA HANDIHALER). Les critères principaux d’évaluation ont été le délai de survenue de la première exacerbation de BPCO, le délai de survenue du décès toutes causes confondues et dans un sous-groupe (906 patients), le VEMS résiduel (pré-dose).
Le délai avant la première exacerbation de BPCO a été numériquement similaire au cours de l’étude entre SPIRIVA RESPIMAT et SPIRIVA HANDIHALER (risque relatif (SPIRIVA RESPIMAT/SPIRIVA HANDIHALER) de 0,98 avec un IC à 95% : 0,93 à 1,03). Le nombre médian de jours avant la première exacerbation a été de 756 jours pour SPIRIVA RESPIMAT et de 719 jours pour SPIRIVA HANDIHALER.
L’effet bronchodilatateur de SPIRIVA RESPIMAT a été maintenu pendant 120 semaines, et a été similaire à celui du SPIRIVA HANDIHALER. La différence moyenne du VEMS résiduel pour SPIRIVA RESPIMAT versus SPIRIVA HANDIHALER a été de -0,010 litres (IC à 95% : -0,038 à 0,018 L).
Dans l’étude clinique TIOSPIR conduite après la mise sur le marché et comparant SPIRIVA RESPIMAT et SPIRIVA HANDIHALER, la mortalité toutes causes confondues (incluant le suivi de la survie) a été similaire (risque relatif SPIRIVA RESPIMAT/SPIRIVA HANDIHALER de 0,96 avec un IC à 95% : 0,84 à 1,09). L’exposition au traitement a été respectivement de 13 135 et de 13 050 patients-années.
Dans les études contrôlées contre placebo mesurant la survie jusqu’à la fin de la période prévue de traitement, le taux de mortalité toutes causes confondues était augmenté dans les groupes traités SPIRIVA RESPIMAT comparativement aux groupes placebo (risque relatif de 1,33 avec un IC à 95% : 0,93 à 1,92), avec une exposition des patients au traitement par SPIRIVA RESPIMAT de 2574 patients-année. L’augmentation de la mortalité a été observée chez des patients ayant des troubles du rythme cardiaque connus. Une diminution de 13% du risque de décès a été observée dans le groupe traité par SPIRIVA HANDIHALER (risque relatif tiotropium/placebo incluant le suivi de la survie: 0,87 avec un IC à 95% : 0,76 à 0,99). L’exposition au traitement par SPIRIVA HANDIHALER a été de 10 927 patients-années. Il n'a pas été mis en évidence d'augmentation du risque de la mortalité dans le sous-groupe de patients présentant des troubles du rythme cardiaque connus ni dans l’étude ayant comparé SPIRIVA HANDIHALER au placebo, ni dans l’étude TIOSPIR ayant comparé SPIRIVA RESPIMAT et SPIRIVA HANDIHALER.
Efficacité et sécurité clinique dans le traitement de l’asthme
Les études de phase III conduites chez les adultes dans l’asthme persistant consistent en deux études d’un an randomisées, en double aveugle, contrôlées contre placebo chez un total de 907 patients asthmatiques (dont 453 traités par SPIRIVA RESPIMAT) traités par l’association d’un corticostéroïde inhalé (≥ 800 µg de budésonide/jour ou équivalent) et d’un bronchodilatateur de longue durée d’action. Les critères de jugement principaux incluaient la mesure de la fonction pulmonaire et les exacerbations sévères.
Études PrimoTinA dans l’asthme
Les deux études cliniques d’un an incluant des patients asthmatiques symptomatiques malgré un traitement continu par au moins l’association d’un CSI (≥ 800 µg de budésonide/jour ou équivalent) et d’un LABA, ont mis en évidence une amélioration cliniquement significative de la fonction pulmonaire dans les groupes de patients où était ajouté un traitement par SPIRIVA RESPIMAT comparativement à l’ajout d’un placebo.
À la semaine 24, les améliorations moyennes des valeurs du VEMS au pic et résiduelle ont été respectivement de 0,110 litre (IC à 95 % : 0,063 à 0,158 litre ; p < 0,0001) et de 0,093 litre (IC à 95 % : 0,050 à 0,137 litre ; p < 0,0001). L’amélioration de la fonction pulmonaire par rapport au placebo a été maintenue pendant 24 heures.
Dans les études PrimoTinA dans l'asthme, le risque d’exacerbations sévères de l’asthme était réduit de 21% dans le groupe de patients symptomatiques traités par CSI, LABA et tiotropium (N = 453) comparativement au groupe de patients traités par CSI, LABA et placebo (N = 454). La réduction du nombre moyen d’exacerbations sévères de l’asthme par patient-année a été de 20%.
Une réduction de 31% du risque de détérioration de l’asthme et une réduction de 24% du nombre moyen de détérioration de l’asthme par patient-année étaient également observées (voir Tableau 2).
Tableau 2 : Exacerbations chez les patients symptomatiques traités par une association de CSI (≥ 800 µg de budésonide/jour ou équivalent) et de LABA (études PrimoTinA dans l’asthme)
|
Étude |
Critère |
SPIRIVA RESPIMAT, en complément d’un traitement par au moins un CSIa/LABA (N = 453) |
Placebo, en complément d’un traitement par au moins un CSIa/LABA (N = 454) |
% de réduction du risque (IC à 95 %) |
Valeur de p |
|
Analyse groupée des 2 études de phase III d’un an |
Nombre de jours avant la 1re exacerbation sévère de l’asthme |
282c |
226c |
21b (0 - 38) |
0,0343 |
|
Nombre moyen d’exacerbations sévères de l’asthme par patient-année |
0,530 |
0,663 |
20d (0 - 36) |
0,0458 |
|
|
Nombre de jours avant la 1re aggravation de l’asthme |
315c |
181c |
31b (18 - 42) |
< 0,0001 |
|
|
Nombre moyen d’aggravations de l’asthme par patient-année |
2,145 |
2,835 |
24d (9 - 37) |
0,0031 |
a ≥800µg de budésonide/jour ou équivalent
b Risque relatif, intervalle de confiance et valeur de p obtenus à partir d’un modèle aléatoire proportionnel de Cox avec uniquement le traitement comme effet. La réduction du risque exprimé en pourcentage est calculée avec la formule suivante : 100 (1 - risque relatif).
c Délai avant le premier événement : nombre de jours sous traitement écoulés avant que 25 % / 50 % des patients présentent au moins une exacerbation sévère de l’asthme/aggravation de l’asthme.
d Le risque relatif a été obtenu par une régression de Poisson pondérée par un exposant logarithmique (en année). La réduction du risque exprimé en pourcentage est calculée avec la formule suivante : 100 (1 - risque relatif).
Population pédiatrique
Bronchopneumopathie chronique obstructive (BPCO)
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec SPIRIVA RESPIMAT dans tous les sous-groupes de la population pédiatrique dans le cadre du traitement de la BPCO (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
Asthme
Toutes les études de phase III conduites dans la population pédiatrique (1-17 ans) dans l’asthme persistant étaient randomisées, en double aveugle et contrôlées contre placebo. Tous les patients recevaient un des traitements de fond incluant un CSI.
Asthme sévère
Adolescents (12-17 ans)
Dans l’étude PensieTinA de 12 semaines conduite dans l’asthme, un total de 392 patients (130 recevant SPIRIVA RESPIMAT) symptomatiques sous une forte dose de CSI avec un traitement de contrôle ou une dose moyenne de CSI avec deux traitements de contrôle ont été inclus.
Pour les patients âgés de 12 à 17 ans, une forte dose de CSI a été définie comme une dose > 800‑1600 μg de budésonide/jour ou équivalent ; une dose moyenne comme une dose de 400‑800 μg de budésonide/jour ou équivalent. De plus, les patients âgés de 12 à 14 ans pouvaient recevoir une dose de CSI > 400 μg de budésonide/jour ou équivalent et au minimum un traitement de contrôle ou une dose de CSI ≥ 200 µg de budésonide/jour ou équivalent et au minimum deux traitements de contrôle.
Dans cette étude, SPIRIVA RESPIMAT a montré des améliorations de la fonction pulmonaire par rapport au placebo quand il était utilisé en traitement d’appoint au traitement de fond, cependant, les différences sur le VEMS au pic et résiduel n’étaient pas statistiquement significatives.
· A la semaine 12, les améliorations moyennes du VEMS au pic et résiduel étaient respectivement de 0,090 litres (IC 95% : -0,019 à 0,198 litres, p=0,1039) et 0,054 litres (IC 95% : -0,061 à 0,168 litres, p=0,3605).
· A la semaine 12, SPIRIVA RESPIMAT a significativement amélioré le DEP du matin et du soir (matin 17,4 L/min ; IC 95% : 5,1 à 29,6 L/min ; soir 17,6 L/min ; IC 95% : 5.9 à 29,6 L/min).
Enfants (6-11 ans)
Dans l’étude VivaTinA de 12 semaines conduite dans l’asthme, un total de 400 patients (130 recevant SPIRIVA RESPIMAT) symptomatiques sous une forte dose de CSI avec un traitement de contrôle ou une dose moyenne de CSI avec deux traitements de contrôle ont été inclus. Une forte dose de CSI a été définie comme une dose > 400 μg de budésonide/jour ou équivalent, une dose moyenne comme une dose de 200‑400 μg de budésonide/jour ou équivalent.
Dans cette étude, SPIRIVA RESPIMAT a montré des améliorations significatives de la fonction pulmonaire par rapport au placebo quand il était utilisé en traitement d’appoint au traitement de fond,
· A la semaine 12, les améliorations moyennes du VEMS au pic et résiduel étaient respectivement de 0,139 litres (IC 95% : 0,075 à 0,203 litres, p < 0,0001) et 0,087 litres (IC 95% : 0,019 à 0,154 litres, p=0,0117).
Asthme modéré
Adolescents (12-17 ans)
Dans l’étude RubaTinA d’un an conduite dans l’asthme sur un total de 397 patients (134 recevant SPIRIVA RESPIMAT) symptomatiques sous une dose moyenne de CSI (200‑800 μg de budésonide/jour ou équivalent pour les patients âgés de 12 à 14 ans ou 400‑800 μg de budésonide/jour ou équivalent pour les patients âgés de 15 à 17 ans), SPIRIVA RESPIMAT a montré des améliorations significatives de la fonction pulmonaire par rapport au placebo quand il était utilisé en traitement d’appoint au traitement de fond,
Enfants (6-11 ans)
Dans l’étude CanoTinA d’un an conduite dans l’asthme sur un total de 401 patients (135 recevant SPIRIVA RESPIMAT) symptomatiques sous une dose moyenne de CSI (200‑400 μg de budésonide/jour ou équivalent), SPIRIVA RESPIMAT a montré des améliorations significatives de la fonction pulmonaire par rapport au placebo quand il était utilisé en traitement d’appoint au traitement de fond.
Enfants (1-5 ans)
Une étude clinique de phase II/III de 12 semaines randomisée, en double-aveugle, contrôlée contre placebo (NinoTinA dans l’asthme) a été menée sur un total de 101 enfants asthmatiques (31 recevant SPIRIVA RESPIMAT) avec des traitements de fond qui incluaient un CSI. Une chambre de retenue valvée Aerochamber Plus Flow-Vu® munie d’un masque a été utilisée pour administrer le médicament à l’essai chez 98 patients.
L’objectif principal de l’essai était la sécurité ; les évaluations de l’efficacité étaient exploratoires.
Le nombre et le pourcentage de patients ayant rapporté des effets indésirables quel que soit le lien de causalité sont présentés dans le tableau 3. Le nombre d’effets indésirables à type d’asthme a été inférieur pour SPIRIVA RESPIMAT comparé au placebo. Les évaluations exploratoires de l’efficacité n’ont pas montré de différences entre SPIRIVA RESPIMAT et le placebo.
Tableau 3 : Fréquence des effets indésirables rapportés chez ≥ 5 patients dans l’étude Nino-TinA dans l’asthme (enfants âgés de 1 à 5 ans)
|
|
Placebo N (%) |
SPIRIVA RESPIMAT N (%) |
|
Nombre de patients |
34 (100,0) |
31 (100,0) |
|
Patients présentant un effet indésirable |
25 (73,5) |
18 (58,1) |
|
Rhinopharyngite |
5 (14,7) |
2 (6,5) |
|
Infection des voies respiratoires supérieures |
1 (2,9) |
5 (16,1) |
|
Asthme* |
10 (29,4) |
2 (6,5) |
|
Pyrexie |
6 (17,6) |
3 (9,7) |
* « Asthme » est le terme préférentiel de MedDRA. Les termes MedDRA de bas niveau étaient « Asthme aggravé » ou « Exacerbation de l’asthme ».
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec SPIRIVA RESPIMAT dans le sous-groupe des patients pédiatriques âgés de moins de 1 an (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
Efficacité et sécurité clinique dans le traitement de la mucoviscidose
Le programme de développement clinique dans la mucoviscidose incluait 3 études multicentriques chez 959 patients âgés de 5 mois et plus. Les patients âgés de moins de 5 ans utilisaient une chambre d’inhalation (AeroChamber Plus®) munie d’un masque et étaient inclus uniquement pour une évaluation de la sécurité. Les deux études pivots (une étude de phase II de recherche de dose et une étude de phase III) comparaient les effets de SPIRIVA RESPIMAT (tiotropium 5 µg : 469 patients) par rapport au placebo (315 patients) sur la fonction pulmonaire (VEMS exprimé en pourcentage des valeurs prédites ASC0-4h (Aire Sous la Courbe), et VEMS résiduel) pendant la période de 12 semaines, randomisée, en double aveugle ; l’étude de phase III incluait de plus une phase d'extension en ouvert d'étude à long terme pouvant aller jusqu’à 12 mois. Dans ces études, tous les traitements à visée respiratoire, à l’exception des anticholinergiques, étaient autorisés comme traitement concomitant, notamment les béta-2 agonistes à longue durée d’action, les mucolytiques et les antibiotiques.
Les effets sur la fonction pulmonaire sont présentés dans le tableau 4. Aucune amélioration significative des symptômes et de l’état de santé (évaluation des exacerbations à l’aide du RSSQ (questionnaire sur les symptômes respiratoires et systémiques) et de la qualité de vie à l’aide du questionnaire de qualité de vie dans la mucoviscidose (CFQ, Cystic Fibrosis Questionnaire)) n’a été observée.
Tableau 4 : Valeurs absolues des différences moyennes des variations après 12 semaines par rapport à la valeur initiale, ajustée en fonction du placebo.
|
|
Phase II |
Phase III |
||||
|
|
Population globale (NSpiriva = 176, Nplacebo = 168) |
Population globale (NSpiriva = 293, Nplacebo = 147) |
Age ≤ 11 ans |
Age ≥ 12 ans |
||
|
(NSpiriva = 95, Nplacebo = 47) |
(NSpiriva = 198, Nplacebo = 100) |
|||||
|
|
moyenne (IC95%) |
Valeur de p |
moyenne (IC95%) |
Valeur de p |
moyenne (IC95%) |
moyenne (IC95%) |
|
VEMS ASC0-4h (% de la valeur théorique)a Valeur absolue de la variation |
3,39 (1,67; 5,12) |
<0,001 |
1,64 (-0,27 ; 3,55) |
0,092 |
-0,63 (-4,58 ; 3,32) |
2,58 (0,50 ; 4,65) |
|
VEMS ASC0-4h (litres) Valeur absolue de la variation |
0,09 (0,05; 0,14) |
<0,001 |
0,07 (0,02; 0,12) |
0,010 |
0,01 (-0,07 ; 0,08) |
0,10 (0,03 ; 0,17) |
|
VEMS résiduel (% de la valeur théorique)a Valeur absolue de la variation |
2,22 (0,38; 4,06) |
0,018 |
1,40 -0,50; 3,30 |
0,150 |
-1,24 (-5,20 ; - 271) |
2,56 (0,49 ; 4,62) |
|
VEMS résiduel (litres) Valeur absolue de la variation |
0,06 (0,01; 0,11) |
0,028 |
0,07 (0,02; 0,12) |
0,012 |
-0,01 (-0,08; 0,06) |
0,10 (0,03; 0,17) |
a Critères d’évaluation principaux
Tous les effets indésirables liés au médicament observés dans les études dans la mucoviscidose sont des effets indésirables connus du tiotropium (voir rubrique 4.8). Les événements indésirables considérés comme liés au médicament les plus fréquemment observés pendant les 12 semaines de traitement en double aveugle étaient la toux (4,1%) et la sécheresse buccale (2,8%).
Le nombre et le pourcentage de patients ayant rapporté des événements indésirables (EI) à prendre en considération spécifiquement dans la mucoviscidose quel que soit le lien de causalité sont présentés dans le tableau 5. Les signes et symptômes considérés comme des manifestations de la mucoviscidose ont augmenté en nombre avec le tiotropium, en particulier chez les patients ≤ 11 ans, bien que la différence ne soit pas statistiquement significative.
Tableau 5 : Pourcentage de patients avec des effets indésirables à prendre en compte spécifiquement dans la mucoviscidose quel que soit le lien de causalité, par groupes d’âge, durant les 12 semaines de traitement (Phase II et Phase III regroupées)
|
|
Age ≤11 ans |
Age ≥12 ans |
||
|
|
Nplacebo=96 |
NSpiriva=158 |
Nplacebo=215 |
NSpiriva=307 |
|
Douleur abdominale |
7,3 |
7,0 |
5,1 |
6,2 |
|
Constipation |
1,0 |
1,9 |
2,3 |
2,6 |
|
Syndrome d’occlusion distale de l‘intestin |
0,0 |
0,0 |
1,4 |
1,3 |
|
Infections de l'appareil respiratoire |
34,4 |
36,7 |
28,4 |
28,3 |
|
Augmentation des expectorations |
1,0 |
5,1 |
5,6 |
6,2 |
|
Exacerbations |
10,4 |
14,6 |
18,6 |
17,9 |
"Syndrome d’occlusion distale de l‘intestin" et "Augmentation des expectorations" sont des termes préférentiels de MedDRA. "Infections de l'appareil respiratoire" est le groupe de terme de haut niveau de MedDRA. "Douleur abdominale", "Constipation" et "Exacerbations" sont un ensemble de termes préférentiels de MedDRA.
Trente quatre (10,9%) des patients randomisés dans le groupe placebo et 56 (12,0%) des patients randomisés dans le groupe SPIRIVA RESPIMAT ont présenté un événement indésirable grave.
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec SPIRIVA RESPIMAT dans le sous-groupe de la population pédiatrique de moins de 1 an.
5.2. Propriétés pharmacocinétiques
Le bromure de tiotropium est un ammonium quaternaire non chiral peu soluble dans l’eau. Il est présenté sous forme de solution à inhaler administrée par l’intermédiaire du dispositif d’inhalation RESPIMAT. Approximativement 40 % de la dose inhalée est déposée dans les poumons, l’organe cible, le reste étant déposé dans le tractus gastro-intestinal. Certaines des données pharmacocinétiques indiquées ci-dessous ont été obtenues avec des doses supérieures à la posologie thérapeutique recommandée.
b) Caractéristiques pharmacocinétiques générales de la substance active après l’administration de la spécialité
Absorption : après inhalation chez des volontaires sains jeunes, les données concernant l’excrétion urinaire suggèrent qu’approximativement 33 % de la dose inhalée atteint la circulation systémique. La biodisponibilité absolue des solutions orales de bromure de tiotropium est de 2 à 3%. La prise d'aliments n’a a priori pas d’influence sur l'absorption de cet ammonium quaternaire.
Les concentrations plasmatiques maximales de tiotropium ont été observées 5 à 7 minutes après l’inhalation.
À l’équilibre, les concentrations plasmatiques maximales de tiotropium chez les patients présentant une BPCO ont été de 10,5 pg/ml et ont diminué rapidement selon un modèle à compartiments multiples. Les concentrations plasmatiques minimales à l’équilibre ont été de 1,60 pg/ml. Après l’administration de la même dose à des patients asthmatiques, la concentration plasmatique maximale de tiotropium à l’état d’équilibre, atteinte en 5 minutes, est de 5,15 pg/ml.
L’exposition systémique au tiotropium après inhalation par l’inhalateur Respimat a été similaire à celle observée après inhalation de tiotropium par le dispositif HandiHaler.
Distribution : la liaison du bromure de tiotropium aux protéines plasmatiques est de 72 % et son volume de distribution est de 32 l/kg. Les concentrations locales pulmonaires ne sont pas connues, mais le mode d'administration laisse penser qu'elles sont beaucoup plus élevées. Les études chez le rat ont montré que le tiotropium ne traverse pas la barrière hémato-encéphalique de façon significative.
Biotransformation : le métabolisme du bromure de tiotropium est faible. Chez de jeunes volontaires sains, l'excrétion urinaire de la substance non métabolisée atteint 74 % de la dose après une administration intraveineuse. L'ester du bromure de tiotropium est clivé, indépendamment d’un mécanisme enzymatique, en un dérivé alcool (N-méthylscopine) et un dérivé acide (acide dithiénylglycolique), inactifs sur les récepteurs muscariniques. Les études réalisées in vitro sur des microsomes hépatiques et des hépatocytes d'origine humaine montrent qu'une petite partie supplémentaire (< 20% de la dose administrée par voie intraveineuse) est métabolisée par une réaction d'oxydation dépendante du cytochrome P450 (CYP) puis par conjugaison avec le glutathion, donnant naissance à une série de métabolites de phase II.
Les études in vitro effectuées sur des microsomes hépatiques suggèrent une inhibition du métabolisme par les inhibiteurs du CYP 2D6 (et 3A4), la quinidine, le kétoconazole et le gestodène. Les iso-enzymes CYP 2D6 et 3A4 sont donc impliquées dans une voie métabolique résultant en l’élimination d’une faible partie de la dose. Il n’a pas été mis en évidence d’effet inhibiteur, même avec des concentrations élevées, sur les iso-enzymes CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 ou 3A sur les microsomes hépatiques humains.
Elimination : la demi-vie réelle du tiotropium est comprise entre 27 et 45 heures après inhalation par des volontaires sains et des patients présentant une BPCO. La demi-vie réelle chez des patients asthmatiques a été de 34 heures. La clairance totale a été de 880 ml/min après une dose intraveineuse chez de jeunes volontaires sains. Le tiotropium administré par voie intraveineuse est essentiellement éliminé par voie urinaire sous forme inchangée (74%). Après inhalation de la solution à l’état d’équilibre par des patients atteints de BPCO, l’excrétion urinaire est de 18,6 % de la dose (0,93 µg), le reste étant principalement constitué de médicament non absorbé dans l’intestin puis éliminé dans les fèces. Après l’inhalation de la solution par des volontaires sains, l’excrétion urinaire est de 20,1 à 29,4 % de la dose, le reste étant principalement constitué de médicament non absorbé dans l’intestin puis éliminé dans les fèces. Après l’administration chez des patients asthmatiques, 11,9 % (0,595 µg) de la dose ont été excrétés sous forme inchangée dans les urines pendant une période de 24 heures à l’état d’équilibre. La clairance rénale du tiotropium est plus élevée que la clairance de la créatinine, reflétant une sécrétion urinaire.
Après l’inhalation quotidienne par des patients présentant une BPCO, l’état d’équilibre pharmacocinétique a été atteint au 7ème jour sans accumulation par la suite.
Linéarité / non-linéarité : le tiotropium montre des propriétés pharmacocinétiques linéaires dans l’intervalle thérapeutique quel que soit la formulation.
c) Caractéristiques pharmacocinétiques dans des populations particulières :
Sujet âgé : comme pour les médicaments excrétés majoritairement par voie rénale, la clairance rénale du tiotropium diminue avec l'âge (347 ml/min chez des patients atteints de BPCO de moins de 65 ans contre 275 ml/min chez des patients atteints de BPCO âgés de 65 ans ou plus. Cela n’a pas été associé à une augmentation des valeurs de l’ASC0-6 ss ou de la Cmax,SS. Il n’a pas été mis en évidence de différence d’exposition systémique au tiotropium en fonction de l’âge chez des patients asthmatiques.
Insuffisance rénale : Après inhalation de la dose quotidienne de tiotropium à l’état d’équilibre chez des patients présentant une BPCO, une insuffisance rénale légère (CLCR 50-80 ml/min) a entraîné des valeurs d’ASC0‑6ss légèrement supérieures (de 1,8 à 30%) et des valeurs de Cmax,SS similaires par rapport aux patients présentant une fonction rénale normale (CLCR > 80 ml/min).
Chez les patients atteints de BPCO et présentant une insuffisance rénale modérée à sévère (CLCR <50 ml/min), l'administration intraveineuse d’une seule dose de tiotropium a entraîné le doublement de l’exposition totale (augmentation de 82% de l’ASC0-4h et de 52% de la Cmax) par rapport aux patients présentant une insuffisance rénale normale, ce qui a été confirmé par la mesure des concentrations plasmatiques après inhalation sous forme de poudre sèche. Il n’a pas été mis en évidence d’augmentation significative de l’exposition systémique après inhalation de tiotropium chez les patients asthmatiques présentant une insuffisance rénale légère (ClCR de 50 à 80 ml/mn) par rapport aux patients présentant une fonction rénale normale.
Insuffisance hépatique : il n’est pas attendu de modification significative de la pharmacocinétique du tiotropium en cas d’insuffisance hépatique, dans la mesure où le produit est essentiellement éliminé par voie rénale (74 % chez le jeune volontaire sain) et métabolisé par simple clivage non enzymatique des liaisons esters en produits pharmacologiquement inactifs.
Patients japonais présentant une BPCO : dans une comparaison croisée, les concentrations plasmatiques maximales moyennes de tiotropium, 10 minutes après l’administration à l’état d’équilibre, étaient de 20% à 70% supérieures chez les patients japonais présentant une BPCO par rapport aux patients caucasiens après inhalation de tiotropium, mais aucun signal d’une mortalité supérieure ou d’un risque cardiaque augmenté n’a été détecté chez les patients japonais. Les données pharmacocinétiques disponibles pour les autres origines ethniques sont insuffisantes.
Pédiatrie :
Asthme
Le pic et l’exposition totale au tiotropium (ASC et excrétion urinaire) sont comparables entre les patients asthmatiques âgés de 6 à 11 ans, ceux âgés de 12 à 17 ans et les patients âgés de 18 ans ou plus. Sur la base de l’excrétion urinaire, l’exposition totale au tiotropium des patients âgés de 1 à 5 ans était de 52 à 60% plus basse que chez les patients plus âgés. Quand elles ont été ajustées à la surface corporelle les données d’exposition totale ont été comparables dans toutes les tranches d’âge. SPIRIVA RESPIMAT a été administré avec une chambre de retenue valvée munie d’un masque chez les patients âgés de 1 à 5 ans.
BPCO
Le programme de développement dans la bronchopneumopathie chronique obstructive (BPCO) ne comprenait pas l'étude des patients pédiatriques (voir rubrique 4.2).
Mucoviscidose
Après une inhalation de 5 µg de tiotropium, la concentration plasmatique de tiotropium chez les patients âgés de 5 ans et plus atteints de mucoviscidose était de 10,1 pg/ml à l’état d’équilibre 5 minutes après l’administration et a ensuite diminué rapidement. La fraction de dose disponible chez les patients de moins de 5 ans atteints de mucoviscidose ayant utilisé la chambre d’inhalation munie d'un masque facial était environ 3 à 4 fois plus faible que celle observée chez les patients atteints de mucoviscidose âgés de 5 ans et plus. Chez les patients de moins de 5 ans atteints de mucoviscidose, le niveau d'exposition systémique au tiotropium était fonction du poids corporel.
d) Relation(s) entre paramètres pharmacocinétiques et paramètres pharmacodynamiques
Il n'y a pas de relation directe entre les paramètres pharmacocinétiques et la pharmacodynamie du produit.
5.3. Données de sécurité préclinique
Chez l'animal, ont ainsi été observées une diminution de la consommation de nourriture, une réduction de la prise de poids, une sécheresse buccale et nasale, une réduction de la sécrétion de larmes et de salive, une mydriase et une augmentation du rythme cardiaque. D'autres effets notables ont été observés lors des études de toxicité en administration répétée : légère irritation du tractus respiratoire chez le rat et la souris, se manifestant par une rhinite et des altérations de l'épithélium de la cavité nasale et du larynx, et prostatite avec dépôts de substances de type protéinique et lithiases vésicales chez le rat.
Les mêmes modifications pharmacologiques directes et indirectes ainsi que des rhinites ont été observées dans les études de toxicité en doses répétées chez les jeunes rats exposés du 7ème jour après la naissance jusqu’à leur maturité sexuelle. Aucune toxicité systémique et aucun effet toxicologique significatif sur les paramètres principaux du développement de la trachée ou des organes vitaux n’ont été constatés.
Des effets délétères sur la gestation, le développement embryo-foetal, la parturition ou le développement post-natal n’ont été observés qu'à des doses toxiques pour les mères.
Le bromure de tiotropium n'a pas induit d'effets tératogènes chez le rat et le lapin. Dans une étude de la reproduction et de la fertilité chez le rat, aucun effet indésirable n’a été observé sur la fertilité ou l'accouplement chez les parents et leur descendance aux doses administrées.
Les effets sur l'appareil respiratoire (irritation) et uro-génital (prostatite), ainsi que des effets délétères sur la reproduction ont été observés après administration locale ou systémique de doses cinq fois supérieures à la dose thérapeutique. Les études de génotoxicité et de carcinogenèse n’ont pas révélé de risque particulier pour l’Homme.
Edétate disodique
Eau purifiée
Acide chlorhydrique à 3,6 % (pour l'ajustement du pH)
3 ans.
Cartouche après insertion de la cartouche dans l'inhalateur : 3 mois.
Inhalateur après insertion de la première cartouche dans l’inhalateur : 1 an.
Limite d’utilisation recommandée : 6 cartouches par inhalateur.
Note : le fonctionnement de l’inhalateur RESPIMAT réutilisable a été vérifié au cours de 540 pressions test (540 bouffées) (correspondant à 9 cartouches).
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
Type et matériel de conditionnement directement en contact avec le produit :
Solution en cartouche de polyéthylène/polypropylène comprenant un bouchon de polypropylène muni d'un joint d'étanchéité intégré en silicone. La cartouche est incluse dans un cylindre d'aluminium.
Chaque cartouche contient 4 ml de solution à inhaler.
Présentations :
Boîte contenant 1 inhalateur RESPIMAT réutilisable et 1 cartouche délivrant 60 bouffées (60 pressions) (soit 30 jours de traitement).
Boîte contenant 1 inhalateur RESPIMAT réutilisable et 3 cartouches délivrant chacune 60 bouffées (60 pressions) (soit 30 jours de traitement).
Boîte de recharge unique : 1 cartouche délivrant 60 bouffées (60 pressions) (soit 30 jours de traitement).
Boîte de recharges triple : 3 cartouches délivrant chacune 60 bouffées (60 pressions) (soit 30 jours de traitement).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
BOEHRINGER INGELHEIM INTERNATIONAL GMBH
BINGER STRASSE 173
55216 INGELHEIM AM RHEIN
ALLEMAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 684 4 8 : 1 cartouche (PE/Polypropylène) de 60 bouffées avec 1 inhalateur réutilisable.
· 34009 301 684 5 5 : 1 cartouche (PE/Polypropylène) de 60 bouffées.
· 34009 301 699 5 7 : 3 cartouches (PE/Polypropylène) de 60 bouffées chacune avec 1 inhalateur réutilisable.
· 34009 301 699 6 4 : 3 cartouches (PE/Polypropylène) de 60 bouffées chacune.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
|
|
| Plan du site | Accessibilité | Contact | Téléchargement | Declaration de confidentialité | Service-Public.fr | Legifrance | Gouvernement.fr |