Dernière mise à jour le 29/06/2026
DEPAKINE 200 mg/ml, solution buvable

: Ce médicament fait l'objet d'une surveillance renforcée. Pour plus d'informations, cliquez ici
Indications thérapeutiques
Classe pharmacothérapeutique ANTIEPILEPTIQUE - code ATC N03AG01
DEPAKINE appartient à une famille de médicaments appelée antiépileptiques.
Ce médicament est utilisé pour le traitement des différentes formes d’épilepsie chez l’adulte et l’enfant. Chez l’enfant, il est également utilisé pour le traitement préventif des convulsions liées à la fièvre.
Présentations
> 1 flacon(s) en verre brun de 40 ml avec mesurette graduée polyéthylène
Code CIP : 302 930-0 ou 34009 302 930 0 3
Déclaration de commercialisation : 19/01/1967
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 3,70 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 4,72 €
- Taux de remboursement :65%
Documents de bon usage du médicament
- Femmes en âge de procréer ayant un trouble bipolaire : spécialités à base de valproate et alternatives médicamenteuses
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Décembre 2018
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 08/06/2016 | Renouvellement d'inscription (CT) | Le service médical rendu de ces spécialités reste important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l\'aide)
Autres informations
- Titulaire de l'autorisation : SANOFI WINTHROP INDUSTRIE
- Conditions de prescription et de délivrance :
- délivrance ne pouvant se faire qu'après vérification de l'attestation co-signée
- liste II
- médicament nécessitant une surveillance particulière pendant le traitement
- pour adolescents de sexe masculin et hommes susceptibles de procréer : prescription initiale réservée à certains spécialistes
- pour enfants et adolescents de sexe féminin, femmes susceptibles de procréer et femmes enceintes : prescription initiale annuelle réservée à certains spécialistes
- prescription nécessitant la signature annuelle par le médecin et la patiente ou le patient d'une attestation d'information
- prescription réservée aux spécialistes et services NEUROLOGIE
- prescription réservée aux spécialistes et services PEDIATRIE
- renouvellement non restreint
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 241 047 2
ANSM - Mis à jour le : 30/10/2025
DEPAKINE 200 mg/ml, solution buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Valproate de sodium............................................................................................................ 20,00 g
Pour 100 ml.
1 ml de solution correspond à 200 mg de valproate de sodium.
Ce médicament contient du sodium (voir rubrique 4.4).
Excipient à effet notoire : sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
4.1. Indications thérapeutiques
Chez l'adulte : soit en monothérapie, soit en association à un autre traitement antiépileptique:
· Traitement des épilepsies généralisées: crises cloniques, toniques, tonico-cloniques, absences, crises myocloniques, atoniques, et syndrome de Lennox-Gastaut.
· Traitement des épilepsies partielles: crises partielles avec ou sans généralisation secondaire.
Chez l'enfant : soit en monothérapie, soit en association à un autre traitement antiépileptique:
· Traitement des épilepsies généralisées: crises cloniques, toniques, tonico-cloniques, absences, crises myocloniques, atoniques, et syndrome de Lennox-Gastaut.
· Traitement des épilepsies partielles: crises partielles avec ou sans généralisation secondaire.
Chez l'enfant :
· Prévention de la récidive de crises après une ou plusieurs convulsions fébriles, présentant les critères de convulsions fébriles compliquées, en absence d'efficacité d'une prophylaxie intermittente par benzodiazépines.
4.2. Posologie et mode d'administration
Enfants de sexe féminin et femmes en âge de procréer
Le traitement par valproate doit être instauré et surveillé par un médecin spécialiste expérimenté dans la prise en charge de l’épilepsie.
Le valproate ne doit pas être utilisé chez les enfants de sexe féminin et les femmes en âge de procréer sauf en cas d’inefficacité ou d’intolérance aux autres traitements. Dans ce cas, le valproate doit être prescrit et dispensé conformément au programme de prévention de la grossesse du valproate (voir rubriques 4.3 et 4.4).
Adolescents et hommes en âge de procréer
Il est recommandé que le traitement par valproate soit instauré et surveillé par un médecin spécialiste expérimenté dans la prise en charge de l’épilepsie (voir rubriques 4.4 et 4.6).
Parmi les formes pharmaceutiques orales, les formes sirop, solution buvable et granulés LP sont particulièrement adaptées à l’administration chez les enfants de moins de 11 ans.
Posologie
Posologie moyenne par 24 heures :
· Nourrissons et enfants : 30 mg par kg (les formes sirop, solution buvable ou granulés à libération prolongée seront de préférence utilisées).
· Adolescents et adultes : 20 à 30 mg par kg (les formes comprimés ou comprimé chrono ou granulés à libération prolongée seront de préférence utilisées).
La prescription doit s’effectuer exclusivement en milligrammes.
Le flacon de solution buvable est accompagné d’une seringue pour administration orale. Les traits de graduation indiquent les doses exprimées en milligrammes (une graduation tous les 25 mg, de 50 mg à 400 mg).
Patients atteints d’insuffisance rénale
Chez les patients atteints d’insuffisance rénale, il peut s’avérer nécessaire de diminuer la posologie, et chez les patients sous hémodialyse, il pourra être nécessaire de l’augmenter. Le valproate de sodium est dialysable (voir la rubrique 4.9). La posologie doit être modifiée selon la surveillance clinique du patient (voir la rubrique 4.4).
Mode d'administration
Voie orale.
Administrer la solution buvable uniquement avec la seringue pour administration orale (piston rose) présente dans la boîte.
La dose quotidienne est à administrer de préférence au cours des repas :
· en 2 prises chez les patients de moins de 1 an,
· en 3 prises chez les patients de plus de 1 an.
La solution sera absorbée après dilution dans un peu de boisson non gazeuse.
Mise en route du traitement
· S'il s'agit d'un malade déjà en traitement et recevant d'autres antiépileptiques, introduire progressivement le valproate de sodium pour atteindre la dose optimum en deux semaines environ, puis réduire éventuellement les thérapeutiques associées en fonction du contrôle obtenu
· S'il s'agit d'un malade ne recevant pas d'autres antiépileptiques, l'ascension de la posologie s'effectue de préférence par paliers successifs tous les 2 ou 3 jours de façon à atteindre la posologie optimum en une semaine environ.
· En cas de nécessité, l'association d'autres antiépileptiques doit être réalisée de manière progressive (voir rubrique 4.5).
· Femmes en âge de procréer, sauf si toutes les conditions du programme de prévention de la grossesse sont remplies (voir rubriques 4.4 et 4.6).
· Antécédent d’hypersensibilité au valproate, au divalproate, au valpromide ou à l'un des excipients mentionnés à la rubrique 6.1.
· Hépatite aiguë.
· Hépatite chronique.
· Antécédent personnel ou familial d’hépatite sévère, notamment médicamenteuse.
· Porphyrie hépatique.
· Patient ayant des troubles connus du cycle de l’urée (voir rubrique 4.4).
· Patient présentant une déficience systémique primaire en carnitine, non corrigée (voir rubrique 4.4 « Patients à risque d’hypocarnitinémie »).
· Le valproate est contre-indiqué chez les patients souffrant de troubles mitochondriaux connus, causés par des mutations du gène nucléaire codant l’enzyme mitochondriale polymérase gamma (POLG), par ex. le syndrome d’Alpers-Huttenlocher, et chez les enfants de moins de deux ans suspectés d’avoir un trouble lié à la POLG (voir la rubrique 4.4).
· Association au millepertuis (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
Mises en garde spéciales
|
Programme de prévention de la grossesse Le valproate est un tératogène puissant entraînant un risque élevé de malformations congénitales et de troubles neuro-développementaux chez les enfants exposés in utero au valproate (voir rubrique 4.6). Le valproate ne doit pas être utilisé chez les enfants de sexe féminin et les femmes en âge de procréer sauf en cas d’inefficacité ou d’intolérance aux autres traitements. Si aucun autre traitement n’est possible, se conformer au programme de prévention de la grossesse ci-après. DEPAKINE est contre-indiqué dans les cas suivants : · Chez les femmes enceintes, sauf en l’absence d’alternative thérapeutique appropriée (voir rubriques 4.3 et 4.6). · Chez les femmes en âge de procréer, sauf si toutes les conditions du programme de prévention de la grossesse sont remplies (voir rubriques 4.3 et 4.6). Conditions du programme de prévention de la grossesse Le prescripteur doit s’assurer que : · les situations individuelles sont évaluées au cas par cas, en impliquant la patiente dans la discussion afin de garantir son engagement, de discuter des options thérapeutiques et de s’assurer qu’elle a compris les risques et les mesures nécessaires pour réduire ces risques ; · le risque de survenue de grossesse est évalué chez toutes les patientes de sexe féminin ; · la patiente a bien compris et pris conscience des risques de malformations congénitales et de troubles neuro-développementaux, y compris l’ampleur de ces risques pour les enfants exposés in utero au valproate ; · la patiente comprend la nécessité d’effectuer un test de grossesse avant le début du traitement et pendant le traitement, en tant que de besoin ; · la patiente a été conseillée en matière de contraception et est capable de se conformer à la nécessité d’utiliser une contraception efficace (pour plus de détails, voir la sous-rubrique « Contraception » de cet encadré), sans interruption, pendant toute la durée du traitement par valproate ; · la patiente comprend la nécessité qu’un médecin spécialiste expérimenté dans la prise en charge de l’épilepsie réévalue régulièrement (au moins chaque année) le traitement ; · la patiente comprend la nécessité de consulter son médecin dès qu’elle envisage une grossesse afin d’en discuter en temps voulu et de recourir à des options thérapeutiques alternatives avant la conception, et ceci avant d’arrêter la contraception ; · la patiente comprend la nécessité de consulter en urgence son médecin en cas de grossesse ; · la patiente a reçu la brochure d’information patiente ; · la patiente a reconnu avoir compris les risques et précautions nécessaires associés à l’utilisation du valproate (attestation d’information partagée). Ces conditions concernent également les femmes qui ne sont pas sexuellement actives, sauf si le prescripteur considère qu’il existe des raisons incontestables indiquant qu’il n’y a aucun risque de grossesse. Enfants de sexe féminin · Les prescripteurs doivent s’assurer que les parents/soignants des enfants de sexe féminin comprennent la nécessité de contacter le médecin spécialiste aussitôt que les premières menstruations surviennent chez l’enfant de sexe féminin qui utilise du valproate. · Le prescripteur doit s’assurer que les parents/soignants des enfants de sexe féminin ayant leurs premières menstruations, reçoivent une information complète sur les risques de malformations congénitales et de troubles neuro-développementaux, y compris l’ampleur de ces risques, pour les enfants exposés au valproate in utero. · Chez les patientes chez lesquelles les premières menstruations sont apparues, le médecin spécialiste prescripteur doit réévaluer annuellement la nécessité du traitement par valproate et envisager l’ensemble des options thérapeutiques alternatives. Si le valproate est le seul traitement approprié, la nécessité d’utiliser une contraception efficace et toutes les autres conditions du programme de prévention de la grossesse doivent être discutées. Tous les efforts doivent être faits par le médecin spécialiste pour passer à un traitement alternatif chez les enfants de sexe féminin, et cela avant la puberté ou l’âge adulte. Test de grossesse Une grossesse doit être exclue avant l’instauration du traitement par valproate. Le traitement par valproate ne doit pas être instauré chez les femmes en âge de procréer sans l’obtention d’un test de grossesse négatif (test de grossesse plasmatique d’une sensibilité d’au moins 25 mUI/mL), confirmé par un professionnel de santé, afin d’éliminer toute possibilité d’utilisation involontaire du produit pendant la grossesse. Ce test de grossesse doit être répété à intervalles réguliers pendant le traitement. Contraception Les femmes en âge de procréer qui reçoivent du valproate doivent utiliser une contraception efficace, sans interruption et pendant toute la durée du traitement par valproate. Ces patientes doivent recevoir une information complète sur la prévention de la grossesse, ainsi que des conseils en matière de contraception si elles n’utilisent pas de contraception efficace. Au moins une méthode de contraception efficace (de préférence une méthode dont l’efficacité ne dépend pas de l’utilisateur, telle qu’un dispositif intra-utérin ou un implant), ou deux méthodes de contraception complémentaires incluant une méthode barrière, doivent être utilisées. Lors du choix de la méthode de contraception, les situations individuelles doivent être examinées au cas par cas, en impliquant la patiente dans la discussion afin de garantir son engagement et son observance des mesures choisies. L’ensemble des conseils relatifs à une contraception efficace doivent être suivis, même en cas d’aménorrhée. Médicaments contenant des œstrogènes Une utilisation concomitante avec des médicaments contenant des œstrogènes, y compris les contraceptifs hormonaux contenant des œstrogènes, peut potentiellement entraîner une diminution de l’efficacité du valproate (voir rubrique 4.5). Les médecins prescripteurs doivent surveiller la réponse clinique (contrôle de l’épilepsie) à l’initiation ou à l’arrêt des médicaments contenant des œstrogènes. A l’inverse, le valproate ne réduit pas l’efficacité des contraceptifs hormonaux. Evaluation annuelle du traitement par un médecin spécialiste Le médecin spécialiste doit réévaluer, au moins chaque année, le traitement par valproate afin de vérifier s’il constitue toujours le traitement le plus approprié pour la patiente. Le médecin spécialiste doit discuter de l’attestation d’information partagée au moment de l’instauration du traitement et lors de chaque évaluation annuelle et doit s’assurer que la patiente a compris son contenu. L’attestation d’information partagée doit être dûment complétée et signée par le prescripteur et la patiente (ou son représentant légal). Planification de grossesse Chez les femmes envisageant une grossesse, un médecin spécialiste expérimenté dans la prise en charge de l’épilepsie doit réévaluer le traitement par valproate et envisager l’ensemble des options thérapeutiques alternatives. Tous les efforts doivent être faits pour passer à un traitement alternatif approprié avant la conception et cela, avant que la contraception ne soit arrêtée (voir rubrique 4.6). Si un changement de traitement est impossible, la patiente devra recevoir des conseils supplémentaires au regard des risques que le valproate présente pour l’enfant à naître, afin de l’aider à prendre une décision éclairée concernant son projet familial. En cas de grossesse En cas de grossesse chez une femme utilisant du valproate, celle-ci doit être immédiatement orientée vers un médecin spécialiste afin de réévaluer le traitement par valproate et d’envisager des options alternatives. Les patientes dont la grossesse a été exposée au valproate ainsi que leurs partenaires doivent être orientés vers un médecin spécialisé ou expérimenté en tératologie pour évaluation et conseil (voir rubrique 4.6). Le pharmacien doit s’assurer que : · la carte patiente est donnée lors de chaque dispensation de valproate et que les patientes comprennent son contenu ; · les patientes sont informées de ne pas arrêter d’elles-mêmes le traitement par valproate et de contacter immédiatement un médecin spécialiste si elles envisagent ou suspectent une grossesse.
Documents d’information Afin d’aider les professionnels de santé et les patientes à éviter toute exposition fœtale au valproate, le titulaire de l’autorisation de mise sur le marché leur fournit des documents d’information visant à renforcer les mises en garde relatives à la tératogénicité (malformations congénitales) et foetotoxicité (troubles neuro-développementaux) du valproate et de délivrer des recommandations aux femmes en âge de procréer concernant l’utilisation de valproate, ainsi que des détails sur le programme de prévention de la grossesse. Une carte patiente et une brochure d’information patiente doivent être fournies à toutes les patientes qui utilisent du valproate. Une attestation d’information partagée doit être utilisée dûment complétée et signée au moment de l’instauration du traitement et lors de chaque réévaluation annuelle du traitement par valproate par le médecin spécialiste et la patiente (ou son représentant légal). |
Utilisation chez les adolescents et hommes en âge de procréer
Une étude observationnelle rétrospective suggère une augmentation du risque de troubles neurodéveloppementaux (TND) chez les enfants dont le père a été traité par valproate dans les 3 mois précédant la conception comparativement à ceux dont le père était traité par lamotrigine ou lévétiracétam (voir rubrique 4.6).
· de la nécessité de mettre en place des mesures contraceptives efficaces pendant le traitement par valproate et au moins trois mois après l’arrêt de celui-ci, y compris pour leur partenaire,
· de ne pas faire de don de sperme pendant le traitement par valproate et au moins trois mois après l’arrêt du valproate traitement (voir rubrique 4.6).
Le traitement par valproate chez les adolescents et hommes en âge de procréer doit être régulièrement réévalué par le prescripteur afin de déterminer si le valproate reste le traitement le plus approprié. Pour les patients de sexe masculin prévoyant de concevoir un enfant, des alternatives thérapeutiques doivent être envisagées et discutées avec les patients. Les situations individuelles doivent être évaluées au cas par cas. Il est recommandé de demander conseil à un médecin spécialisé dans la prise en charge de l’épilepsie.
Des documents de réduction des risques sont disponibles pour les professionnels de santé ainsi que pour les adolescents et hommes en âge de procréer.
Une attestation d’information partagée doit être utilisée, dûment complétée et signée au moment de l’instauration du traitement par le médecin spécialiste et lors de chaque réévaluation annuelle du traitement par valproate par tout médecin et le patient (ou son représentant légal).
Une brochure d’information patient doit être fourni aux adolescents et hommes en âge de procréer utilisant du valproate.
Convulsions aggravées
Comme avec les autres antiépileptiques, la prise de valproate peut être suivie, au lieu d’une amélioration, d’une aggravation réversible de la fréquence et de la sévérité des convulsions (incluant l’état de mal épileptique) ou de l’apparition de nouveaux types de convulsions chez le patient. En cas de convulsions aggravées, il doit être recommandé au patient de consulter immédiatement son médecin (voir rubrique 4.8). Ces convulsions sont à distinguer de celles qui peuvent survenir lors d'une interaction pharmacocinétique (voir rubrique 4.5), d'une toxicité (hépatopathie ou encéphalopathie - voir rubriques 4.4 et 4.8) ou d'un surdosage.
Ce médicament se transformant dans l'organisme en acide valproïque, il convient de ne pas l'associer à d'autres médicaments subissant cette même transformation afin d'éviter un surdosage en acide valproïque (par exemple : divalproate, valpromide).
Atteintes hépatiques graves
Conditions de survenue
Des atteintes hépatiques d'évolution sévère parfois mortelle ont été rapportées exceptionnellement.
Les nourrissons et les jeunes enfants de moins de 3 ans présentant une épilepsie sévère et notamment une épilepsie associée à des lésions cérébrales, un retard psychique et (ou) une maladie métabolique congénitale, incluant des troubles mitochondriaux tels qu’une déficience en carnitine, des troubles du cycle de l’urée, des mutations POLG (voir rubriques 4.3 et 4.4) ou une maladie dégénérative d'origine génétique, sont les plus exposés à ce risque. Au-delà de l'âge de 3 ans, l'incidence de survenue diminue de façon significative et décroît progressivement avec l'âge.
Dans la grande majorité des cas, ces atteintes hépatiques ont été observées pendant les 6 premiers mois de traitement, le plus souvent entre la 2e et la 12e semaine et, généralement au cours de polythérapie antiépileptique.
Signes évocateurs
Le diagnostic précoce reste avant tout basé sur la clinique. En particulier, il convient de prendre en considération notamment chez les patients à risque (voir conditions de survenue) 2 types de manifestations qui peuvent précéder l'ictère :
· d'une part des signes généraux non spécifiques, généralement d'apparitions soudaines tels qu'asthénie, anorexie, abattement, somnolence, accompagnés parfois de vomissements répétés et de douleurs abdominales,
· d'autre part, une réapparition des crises épileptiques alors que le traitement est correctement suivi.
Il est recommandé d'informer le patient, ou sa famille s'il s'agit d'un enfant, que l'apparition d'un tel tableau doit motiver aussitôt une consultation. Celle-ci comportera, outre l'examen clinique, la pratique immédiate d'un contrôle biologique des fonctions hépatiques.
Détection
Une surveillance des fonctions hépatiques doit être effectuée avant le début du traitement et régulièrement pendant les 6 premiers mois du traitement, en particulier chez les patients à risque.
En cas de modification des traitements associés connus pour leur toxicité hépatique (augmentation de la dose ou nouveau traitement), la surveillance hépatique biologique doit être mise en place de nouveau (voir également la rubrique 4.5 sur le risque d'atteinte hépatique avec les dérivés salicylés, les autres anticonvulsivants incluant le cannabidiol).
Parmi les examens classiques, les tests reflétant la synthèse protéique et notamment le TP (taux de prothrombine) sont les plus pertinents. La confirmation d'un taux de prothrombine anormalement bas, surtout s'il s'accompagne d'autres anomalies biologiques (diminution significative du fibrinogène et des facteurs de coagulation, augmentation de la bilirubine, élévation des transaminases - voir rubrique « Précautions d’emploi »), doit conduire à arrêter le traitement par ce médicament (ainsi que par prudence et s'ils sont co-prescrits, les dérivés salicylés, puisqu'ils utilisent la même voie métabolique).
Pancréatite
Des cas de pancréatites dont l'évolution est parfois mortelle ont été très rarement rapportés. Ils peuvent s'observer quels que soient l'âge et l'ancienneté du traitement, les jeunes enfants paraissant particulièrement exposés à ce risque.
Les pancréatites d'évolution défavorable sont généralement observées chez le jeune enfant, ou chez les patients présentant une épilepsie sévère, des lésions cérébrales ou une polythérapie antiépileptique.
Une insuffisance hépatique associée à la pancréatite augmente le risque d'évolution mortelle.
En cas de syndrome douloureux abdominal aigu comme en cas de manifestations digestives à type de nausées, vomissements et/ou anorexie, il faut savoir évoquer le diagnostic de pancréatite et en cas d'élévations des enzymes pancréatiques, interrompre le traitement en mettant en place les mesures thérapeutiques alternatives qui s'imposent.
Idées et comportements suicidaires
Des idées et comportements suicidaires ont été rapportés chez des patients traités par des antiépileptiques dans plusieurs indications. Une méta-analyse d'essais randomisés, contrôlés versus placebo portant sur des antiépileptiques a également montré une légère augmentation du risque d'idées et de comportements suicidaires. Les causes de ce risque ne sont pas connues et les données disponibles n'excluent pas la possibilité d'une augmentation de ce risque pour le valproate.
Par conséquent les patients doivent être étroitement surveillés pour tout signe d'idées et de comportements suicidaires et un traitement approprié doit être envisagé. Il doit être recommandé aux patients (et leur personnel soignant) de demander un avis médical en cas de survenue de signes d'idées et de comportements suicidaires.
Patients présentant une maladie mitochondriale connue ou suspectée
Le valproate peut déclencher ou aggraver des signes cliniques de la maladie mitochondriale sous-jacente causée par des mutations de l’ADN mitochondrial ainsi que du gène nucléaire codant l’enzyme mitochondriale polymérase γ (POLG).
Notamment, des cas d’insuffisance hépatique aiguë induite par le valproate et des décès liés ont été signalés à un taux plus élevé chez les patients présentant des syndromes héréditaires neurométaboliques causés par des mutations du gène POLG par ex. le syndrome d’Alpers-Huttenlocher.
Des troubles liés à la POLG devraient être soupçonnés chez les patients présentant des antécédents familiaux ou des symptômes évoquant un trouble lié à la POLG, y compris, entre autres, une encéphalopathie inexpliquée, une épilepsie réfractaire (focale, myoclonique), un état de mal épileptique à la présentation, des retards développementaux, une régression psychomotrice, une neuropathie axonale sensitivo-motrice, une myopathie, une ataxie cérébelleuse, une ophthalmoplégie, ou une migraine compliquée avec aura occipitale. Pour une évaluation diagnostique de tels troubles, un test des mutations de la POLG devrait être effectué, conformément à la pratique clinique actuelle (voir la rubrique 4.3).
Réactions indésirables cutanées sévères et angioœdème
Des réactions indésirables cutanées sévères (SCAR) telles que le syndrome de Stevens-Johnson (SSJ), la nécrolyse épidermique toxique (NET) et le syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), un érythème polymorphe et un angioœdème ont été rapportées en lien avec un traitement par valproate. Les patients doivent être informés des signes et symptômes de manifestations cutanées graves et doivent être surveillés étroitement. Si des signes de réactions indésirables cutanées sévères ou angioœdème sont observés, une évaluation rapide est nécessaire et le traitement doit être interrompu si le diagnostic de réactions indésirables cutanées sévères ou angioœdème est confirmé.
Interactions médicamenteuses
La prise de ce médicament est déconseillée en association à la lamotrigine et/ou aux pénems (carbapénèmes) (voir rubrique 4.5).
Troubles cognitifs ou extrapyramidaux
Des troubles cognitifs ou extrapyramidaux peuvent être associés à une imagerie d’atrophie cérébrale. Un tel tableau clinique peut ainsi être confondu avec une pathologie de type démence ou maladie de Parkinson. Ces troubles sont réversibles à l’arrêt du traitement (voir rubrique 4.8).
Information liée à la présence de sodium
Ce médicament contient 28 mg de sodium pour 200 mg de valproate de sodium, ce qui équivaut à 1,4 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte. En tenir compte chez les patients suivant un régime hyposodé strict.
Précautions d’emploi
Pratiquer un contrôle biologique des fonctions hépatiques avant le début du traitement (voir rubrique 4.3) puis une surveillance périodique pendant les 6 premiers mois, tout spécialement chez les patients à risque (voir rubrique 4.4 « Atteintes hépatiques graves - Détection »).
Il est à souligner que comme avec la plupart des antiépileptiques on peut observer, notamment en début de traitement, une augmentation modérée, isolée et transitoire des transaminases, en l’absence de tout signe clinique.
Dans ce cas, il est conseillé de pratiquer un bilan biologique plus complet (en particulier taux de prothrombine), de reconsidérer éventuellement la posologie et de réitérer les contrôles en fonction de l’évolution des paramètres.
Un examen hématologique (NFS incluant les plaquettes, temps de saignement et bilan de coagulation) est recommandé préalablement au traitement, puis à 15 jours et en fin de traitement, ainsi qu’avant une intervention chirurgicale et en cas d’hématomes ou de saignements spontanés (voir rubrique 4.8).
Chez l'insuffisant rénal, il convient de tenir compte de l'augmentation des concentrations sériques libres en acide valproïque et de diminuer la posologie en conséquence.
Troubles du cycle de l'urée et risque d’hyperammoniémie
Ce médicament est contre-indiqué chez les patients porteurs d'un déficit enzymatique du cycle de l'urée. Lorsqu'un déficit enzymatique du cycle de l'urée est suspecté, des examens métaboliques doivent être effectués avant le traitement en raison du risque d'hyperammoniémie avec le valproate. Quelques cas d'hyperammoniémie associée à un état stuporeux ou à un coma ont été décrits chez ces patients (voir rubrique 4.3 et 4.4 « Patients à risque d’hypocarnitinémie et Atteinte sévère du foie »).
Patients à risque d’hypocarnitinémie
L’administration du valproate peut déclencher la survenue ou l’aggravation d’une hypocarnitinémie pouvant entraîner une hyperammoniémie (susceptible de causer une encéphalopathie hyperammoniémique).
D’autres symptômes tels qu’une toxicité hépatique, une hypoglycémie hypocétosique, une myopathie incluant une cardiomyopathie, une rhabdomyolyse, un syndrome de Fanconi ont été observés, principalement chez les patients présentant des facteurs de risque d’hypocarnitinémie ou une hypocarnitinémie préexistante. Les patients présentant un risque accru d’hypocarnitinémie symptomatique lorsqu’ils sont traités par valproate sont les patients présentant des troubles métaboliques, y compris des troubles mitochondriaux liés à la carnitine (voir également la rubrique 4.4 Patients présentant une maladie mitochondriale connue ou suspectée et Troubles du cycle de l’urée et risque d’hyperammoniémie), les patients ayant une altération de l’apport nutritionnel en carnitine, les patients de moins de 10 ans et les patients traités de manière concomitante par des médicaments conjugués au pivalate ou par d’autres antiépileptiques.
Les patients doivent être avertis qu’ils doivent signaler immédiatement tout signe d’hyperammoniémie, tel qu’une ataxie, une altération de la conscience, des vomissements. Une supplémentation en carnitine doit être envisagée lorsque des symptômes d’hypocarnitinémie sont observés.
Les patients présentant une déficience systémique primaire en carnitine et une hypocarnitinémie corrigée ne peuvent être traités par valproate que si les bénéfices du traitement par valproate sont supérieurs aux risques encourus pour ces patients et en l’absence d’une alternative thérapeutique. Chez ces patients, une surveillance de la carnitine doit être mise en place.
Les patients présentant une déficience sous-jacente en carnitine palmitoyltransférase (CPT) de type II doivent être avertis du risque accru de rhabdomyolyse lors de la prise de valproate. Une supplémentation en carnitine doit être envisagée chez ces patients. Voir aussi les rubriques 4.5, 4.8 et 4.9.
Bien que ce médicament soit reconnu comme n’entraînant qu’exceptionnellement des manifestations d’ordre immunologique, son utilisation chez un sujet présentant un lupus érythémateux disséminé devra être pesée en fonction du rapport bénéfice/risque.
A l’instauration du traitement, les patients doivent être informés du risque de prise de poids et des mesures appropriées, essentiellement diététiques, qui doivent être adoptées pour minimiser celle-ci.
L’excrétion du valproate est essentiellement urinaire, en partie sous forme de corps cétoniques, la recherche de cétonurie peut donner des faux positifs chez les patients diabétiques.
La prise d’alcool est déconseillée pendant la durée du traitement par DEPAKINE.
Enfants
Chez l’enfant de moins de 3 ans, il est recommandé de n’utiliser le valproate qu’en monothérapie, après avoir évalué l’intérêt thérapeutique par rapport au risque d’hépatopathie et de pancréatite chez les patients de cette classe d’âge avant l’instauration du traitement (voir rubrique 4.4 « Atteintes hépatiques graves » et également rubrique 4.5).
Chez l'enfant de moins de 3 ans, éviter la prescription simultanée de dérivés salicylés compte tenu du risque d'hépatotoxicité (voir également rubrique 4.4) et du risque hémorragique.
Chez les enfants présentant des antécédents hépato-digestifs inexpliqués (anorexie, vomissements, accès de cytolyse), accès de léthargie ou coma, retard mental ou en cas d'antécédents familiaux de décès néonatals ou dans l'enfance, des explorations métaboliques et notamment une ammoniémie à jeun et post-prandiale doivent être effectuées avant tout traitement par le valproate.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
+ Millepertuis
Risque de diminution des concentrations plasmatiques et de l'efficacité de l'anticonvulsivant.
Associations déconseillées
+ Lamotrigine
Risque majoré des réactions cutanées graves (Syndrome de Lyell).
Par ailleurs, augmentation des concentrations plasmatiques de lamotrigine (diminution de son métabolisme hépatique par le valproate de sodium).
Si l'association s'avère nécessaire, surveillance clinique étroite.
+ Pénems (carbapénèmes)
Risque de survenue de crises convulsives, par diminution rapide des concentrations plasmatiques de l’acide valproïque, pouvant devenir indétectables.
L’administration d’acide valproïque en association à des carbapénèmes a entraîné une diminution des concentrations plasmatiques d'acide valproïque de l’ordre de 60 à 100 % en environ deux jours. En raison de l'apparition rapide et de l'importance de la diminution des concentrations plasmatiques, l’administration simultanée de carbapénèmes chez des patients stabilisés sous acide valproïque ne pouvant être surveillée doit donc être évitée (voir rubrique 4.4).
Associations faisant l'objet de précautions d’emploi
+ Acétazolamide
Augmentation de l’hyperammoniémie, avec risque accru d’encéphalopathie.
Surveillance clinique et biologique régulière.
+ Aztreonam
Risque de survenue de crises convulsives, par diminution des concentrations plasmatiques de l'acide valproïque.
Surveillance clinique, dosages plasmatiques et adaptation éventuelle de la posologie de l'anticonvulsivant pendant le traitement par l'anti-infectieux et après son arrêt.
+ Carbamazépine
Augmentation des concentrations plasmatiques du métabolite actif de la carbamazépine avec signes de surdosage. De plus, diminution des concentrations plasmatiques d'acide valproïque par augmentation de son métabolisme hépatique par la carbamazépine.
Surveillance clinique, dosages plasmatiques et adaptation des posologies des deux anticonvulsivants.
Le traitement concomitant par valproate et clozapine peut augmenter le risque de neutropénie et de myocardite induite par la clozapine. Si l’utilisation concomitante de valproate et de clozapine est nécessaire, une surveillance attentive de ces deux effets est nécessaire.
+ Felbamate
Augmentation des concentrations plasmatiques de l’acide valproïque, avec risque de surdosage.
Surveillance clinique, contrôle biologique et adaptation éventuelle de la posologie du valproate pendant le traitement par le felbamate et après son arrêt.
+ Médicaments contenant des œstrogènes, y compris les contraceptifs hormonaux contenant des œstrogènes
Les œstrogènes sont des inducteurs des isoformes de l’UDP-Glucuronosyl Transférase (UGT) impliquées dans la glucuro-conjugaison du valproate et peuvent augmenter sa clairance ; ceci pourrait entraîner une diminution de la concentration sérique du valproate et potentiellement une diminution de son efficacité (voir rubrique 4.4). Envisager une surveillance des concentrations sériques du valproate.
A l’inverse, en raison de l'absence d'effet inducteur enzymatique, le valproate ne diminue pas l'efficacité des estroprogestatifs chez les femmes sous contraception hormonale.
+ Métamizole
Surveillance de la réponse clinique (contrôle des crises ou contrôle de l'humeur) et envisager la surveillance des concentrations sériques de valproate, le cas échéant.
+ Méthotrexate
Certains cas décrivent une diminution significative des taux sériques de valproate après l’administration de méthotrexate, avec survenue de convulsions.
Les prescripteurs doivent surveiller la réponse clinique (contrôle des convulsions ou contrôle de l’humeur) et envisager une surveillance appropriée des taux sériques de valproate.
+ Nimodipine (voie orale et par extrapolation, voie injectable)
Risque d’augmentation des concentrations plasmatiques de la nimodipine de 50 %. Par conséquent, la posologie de la nimodipine doit être réduite en cas d’hypotension.
+ Phénobarbital, et par extrapolation primidone
Augmentation de l’hyperammoniémie, avec risque accru d’encéphalopathie.
Surveillance clinique et biologique régulière.
+ Phénytoïne, et par extrapolation fosphénytoïne
Augmentation de l’hyperammoniémie, avec risque accru d’encéphalopathie.
Surveillance clinique et biologique régulière.
+ Propofol
Possible augmentation des concentrations sanguines de propofol. Une réduction de la dose de propofol est à envisager en cas d’association avec le valproate.
+ Rifampicine
Risque de survenue de crises convulsives, par augmentation du métabolisme hépatique du valproate par la rifampicine.
Surveillance clinique et biologique et adaptation éventuelle de la posologie de l'anticonvulsivant pendant le traitement par rifampicine et après son arrêt.
+ Rufinamide
Possible augmentation des concentrations de rufinamide, notamment chez l’enfant de moins de 30 kg.
Chez l’enfant de moins de 30 kg : ne pas dépasser la dose totale de 600 mg/j après la période de titration.
+ Topiramate
Augmentation de l’hyperammoniémie, avec risque accru d’encéphalopathie.
Surveillance clinique et biologique régulière.
+ Zidovudine
Risque d'augmentation des effets indésirables, notamment hématologiques, de la zidovudine par diminution de son métabolisme par l'acide valproïque.
Surveillance clinique et biologique régulière. Un hémogramme à la recherche d'une anémie devrait être réalisé au cours des deux premiers mois de l'association.
+ Zonisamide
Augmentation de l’hyperammoniémie, avec risque accru d’encéphalopathie.
Surveillance clinique et biologique régulière.
Autres formes d'interactions
+ Lithium
DEPAKINE n’a pas d’effet sur la lithémie.
+ Risque d’atteintes hépatiques
L'utilisation concomitante de dérivés salicylés doit être évitée chez les enfants de moins de 3 ans en raison du risque de toxicité hépatique (voir rubrique 4.4).
L'utilisation simultanée de valproate et d’autres anticonvulsivants augmente le risque d'atteinte hépatique, en particulier chez les jeunes enfants (voir rubrique 4.4).
L'utilisation en association à du cannabidiol accroit l'incidence de l'augmentation des transaminases. Dans des essais cliniques chez des patients de tout âge recevant simultanément du cannabidiol à des doses de 10 à 25 mg/kg et du valproate, une augmentation des ALAT de plus de 3 fois la limite supérieure de la normale a été rapportée chez 19 % des patients. Une surveillance hépatique adaptée doit être effectuée lorsque le valproate est utilisé en association à d'autres anticonvulsivants potentiellement hépatotoxiques, y compris le cannabidiol. Des réductions de dose ou un arrêt du traitement doivent être envisagés en cas d'anomalies significatives des paramètres hépatiques (voir rubrique 4.4).
+ Médicaments conjugués au pivalate
L’administration concomitante de valproate et de médicaments conjugués au pivalate (tels que le cefditoren pivoxil, l’adéfovir dipivoxil, le pivmecillinam et la pivampicilline) doit être évitée en raison du risque accru de diminution de la carnitine (voir la rubrique 4.4 Patients à risque d’hypocarnitinémie). Les patients pour l’administration concomitante ne peut être évitée doivent faire l’objet d’une surveillance étroite afin de détecter tout signe ou symptôme d’hypocarnitinémie.
4.6. Fertilité, grossesse et allaitement
Le valproate est contre-indiqué (voir rubriques 4.3 et 4.4) :
· pendant la grossesse, sauf en l’absence d’alternative thérapeutique appropriée ;
· chez les femmes en âge de procréer, sauf si toutes les conditions du programme de prévention de la grossesse sont remplies.
Utilisation chez les adolescents et hommes en âge de procréer : voir rubrique 4.4 « mise en garde » et le paragraphe « Adolescents et hommes en âge de procréer » ci-après.
Grossesse et femmes en âge de procréer
Tératogénicité et effets neuro-développementaux suite à une exposition in utero
Chez les femmes, l’utilisation du valproate, qu'il soit en monothérapie ou en polythérapie avec d’autres antiépileptiques, est fréquemment associée à des issues de grossesses anormales. Les données disponibles montrent un risque accru de malformations congénitales majeures et de troubles neuro-développementaux, à la fois en monothérapie et en polythérapie avec le valproate, par rapport à la population non exposée au valproate. Il a été montré que le valproate traverse la barrière placentaire chez l’animal et chez l’Homme (voir rubrique 5.2).
Chez l’animal, des effets tératogènes ont été démontrés chez la souris, le rat et le lapin (voir rubrique 5.3). Dans les études précliniques, une diminution dose-dépendante du poids fœtal a été démontrée chez les animaux exposés au valproate in utero en comparaison aux animaux non exposés (voir rubrique 5.3).
· Malformations congénitales suite à une exposition in utero
Une méta-analyse (incluant des registres et des études de cohortes) a montré qu’environ 11 % des enfants nés de mères épileptiques traitées par le valproate en monothérapie pendant leur grossesse avaient des malformations congénitales majeures. Ceci est supérieur au risque de malformations majeures rencontré dans la population générale (environ 2-3 %).
Le risque de malformations congénitales majeures chez les enfants exposés in utero à une polythérapie d’antiépileptiques incluant le valproate est plus élevé que celui lié à une polythérapie d’antiépileptiques sans le valproate.
Ce risque est dose-dépendant lors d’une monothérapie avec le valproate et les données disponibles suggèrent qu'il est dose-dépendant lors d’une polythérapie avec le valproate. Cependant, aucune dose seuil excluant ce risque n’a pu être déterminée.
Les données disponibles montrent une incidence accrue de malformations mineures et majeures. Les malformations le plus souvent rencontrées incluent des anomalies de fermeture du tube neural (de l’ordre de 2 à 3 %), des dysmorphies faciales, des fentes labiales et fentes palatines, des craniosténoses, des malformations cardiaques, rénales et uro-génitales (notamment hypospadias), des malformations des membres (notamment aplasie bilatérale du radius) et des syndromes polymalformatifs touchant diverses parties du corps.
L’exposition in utero au valproate peut également entraîner un déficit auditif ou une surdité due aux malformations de l’oreille et/ou du nez (effet secondaire) et/ou à la toxicité directe sur la fonction auditive. Les cas décrivent une surdité ou un déficit auditif unilatéral(e) et bilatéral(e). Les évolutions n’ont pas été rapportées pour tous les cas. Lorsque les évolutions sont rapportées, il n’y pas eu de rétablissement dans la majorité des cas.
· Troubles neuro-développementaux suite à une exposition in utero
Les études mettent en évidence que le valproate entraîne un risque accru des troubles neuro- développementaux chez les enfants exposés in utero. Le risque de troubles neuro- développementaux (y compris l'autisme) semble dose-dépendant lorsque le valproate est utilisé en monothérapie mais les données disponibles ne permettent pas de déterminer une dose excluant ce risque.
Lorsque le valproate est administré en polythérapie avec d'autres médicaments antiépileptiques pendant la grossesse, les risques de troubles neuro-développementaux chez l’enfant ont également été significativement augmentés par rapport à ceux de la population générale ou ceux nés de mères épileptiques non traitées.
La période à risque de ces effets est incertaine et la possibilité d'un risque pendant toute la grossesse ne peut être exclue.
Lorsque le valproate est administré en monothérapie, les études menées chez des enfants d'âge préscolaire exposés in utero au valproate montrent que jusqu'à 30 à 40 % d'entre eux présentent des retards de développement dans la petite enfance, tels que des retards dans l’acquisition de la parole et de la marche, des capacités intellectuelles diminuées, des capacités verbales (parole et compréhension) diminuées ainsi que des troubles de la mémoire.
Le quotient intellectuel (QI) mesuré chez des enfants d'âge scolaire (6 ans) exposés in utero au valproate est en moyenne de 7 à 10 points inférieur à celui des enfants exposés à d'autres antiépileptiques. Bien que le rôle des facteurs confondants ne puisse être exclu, il est prouvé que cette diminution de QI observée chez les enfants exposés in utero est indépendante du QI maternel.
Les données sur l’évolution de ces troubles à long terme sont limitées.
Les données disponibles provenant d’une étude basée sur la population montrent que les enfants exposés in utero au valproate ont un risque accru de présenter des troubles du spectre de l’autisme (environ 3 fois plus fréquent) et d'autisme infantile (environ 5 fois plus fréquent), par rapport à la population non exposée dans l'étude.
Des données disponibles provenant d’une autre étude basée sur la population montrent que les enfants exposés in utero au valproate ont un risque accru de développer le trouble du déficit de l’attention/hyperactivité (TDAH) (environ 1,5 fois plus fréquent), par rapport à la population non exposée dans l'étude.
· Nouveau-né petit pour son âge gestationnel (PAG) suite à une exposition in utero
Certaines études épidémiologiques ont suggéré un risque plus élevé de naître avec un petit poids pour l'âge gestationnel (PAG défini comme un poids de naissance inférieur au 10e percentile ajusté pour l'âge gestationnel, stratifié par sexe) pour les enfants exposés au valproate in utero par rapport aux enfants non exposés ou exposés à la lamotrigine. Un PAG a été observé chez environ 11-15% des enfants exposés au valproate in utero, 8-9% des enfants exposés à la lamotrigine, et 5-10% des enfants non exposés.
Les données disponibles chez l'homme ne permettent pas de conclure à un potentiel effet dose-dépendant.
Femmes en âge de procréer
Chez les femmes en âge de procréer, le traitement par DEPAKINE ne doit pas être utilisé sauf en cas d’inefficacité ou d’intolérance aux autres traitements. Si aucun autre traitement n’est possible, DEPAKINE ne peut être instauré qu’à condition de respecter le programme de prévention de la grossesse (voir rubrique 4.4), notamment :
· qu’elles ne soient pas enceintes (test de grossesse plasmatique d’une sensibilité d’au moins 25 mUI/ml négatif à l’instauration du traitement et à intervalles réguliers pendant le traitement) ;
· qu’elles utilisent au moins une méthode de contraception efficace ;
· et qu’elles soient informées des risques liés à l’utilisation de valproate pendant la grossesse.
Chez ces femmes, le rapport bénéfice-risque doit être réévalué attentivement et à intervalles réguliers au cours du traitement (au moins annuellement).
Médicaments contenant des œstrogènes :
Les médicaments contenant des œstrogènes, y compris les contraceptifs hormonaux contenant des œstrogènes, peuvent augmenter la clairance du valproate, ce qui pourrait entraîner une diminution de la concentration sérique du valproate et potentiellement une diminution de son efficacité (voir rubriques 4.4 et 4.5).
Si une grossesse est envisagée
Chez les femmes envisageant une grossesse, un médecin spécialiste expérimenté dans la prise en charge de l’épilepsie doit réévaluer le traitement par valproate et envisager l’ensemble des options thérapeutiques alternatives. Tous les efforts doivent être faits pour passer à un traitement alternatif approprié avant la conception et cela, avant que la contraception soit arrêtée (voir rubrique 4.4). Si un changement de traitement est impossible, la patiente devra recevoir des conseils supplémentaires au regard des risques que le valproate présente pour l’enfant à naître afin de l’aider à prendre une décision éclairée concernant son projet familial.
· Une supplémentation en acide folique avant la grossesse et en début de grossesse pourrait diminuer le risque d’apparition d'anomalies du tube neural inhérent à toute grossesse. A titre d’information, les données disponibles ne mettent pas en évidence d’action préventive de l’acide folique sur les malformations liées au valproate.
Femmes enceintes
Le valproate utilisé dans le traitement de l’épilepsie est contre-indiqué pendant la grossesse, sauf en l’absence d’alternative thérapeutique appropriée (voir rubriques 4.3 et 4.4).
En cas de grossesse chez une femme utilisant du valproate, celle-ci doit être immédiatement orientée vers un médecin spécialiste afin d’envisager l’ensemble des options thérapeutiques alternatives. Pendant la grossesse, les crises tonico-cloniques et l'état de mal épileptique avec hypoxie chez la mère peuvent entraîner des conséquences graves voire fatales pour la mère et l’enfant à naître.
Si, en cas de situations exceptionnelles, malgré les risques connus associés à l’utilisation de valproate pendant la grossesse, et après évaluation attentive des traitements alternatifs, le valproate devait absolument être maintenu pour contrôler l’épilepsie chez une femme enceinte :
· il est indispensable d’utiliser la dose minimale efficace,
· il est recommandé de répartir la posologie quotidienne en plusieurs doses plus petites au cours de la journée. L’utilisation d’une formulation à libération prolongée pourrait être préférable aux autres formulations afin d’éviter les pics plasmatiques (voir rubrique 4.2).
Toutes les patientes dont la grossesse a été exposée au valproate ainsi que leurs partenaires doivent être orientés vers un médecin spécialisé ou expérimenté en tératologie pour évaluation et recevoir des conseils concernant la grossesse exposée :
· une surveillance prénatale spécialisée doit être instaurée en vue de détecter d’éventuelles anomalies touchant le tube neural ou d’autres malformations ;
Avant l’accouchement
Pratiquer un bilan de coagulation comprenant notamment une numération plaquettaire, un dosage du fibrinogène et un temps de coagulation (Temps de Céphaline Activée : TCA) chez la mère avant l'accouchement.
Risque chez le nouveau-né
· De très rares cas de syndrome hémorragique ont été rapportés chez les nouveau-nés de mères traitées par valproate pendant la grossesse. Ce syndrome hémorragique est lié à une thrombopénie, une hypofibrinogénémie et/ou une diminution des autres facteurs de coagulation. Une afibrinogénémie a également été rapportée et peut être fatale. Toutefois, ce syndrome doit être distingué du déficit en facteurs de la vitamine K induit par le phénobarbital et les inducteurs enzymatiques. Un bilan d’hémostase normal chez la mère ne permet pas d'éliminer des anomalies de l'hémostase chez le nouveau-né. Par conséquent, à la naissance, un bilan comprenant une numération plaquettaire, un dosage du fibrinogène, les tests et les facteurs de coagulation sera pratiqué chez les nouveau-nés.
· Des cas d'hypoglycémie ont été rapportés chez des nouveau-nés de mères traitées avec du valproate au cours du troisième trimestre de leur grossesse.
· Des cas d'hypothyroïdie ont été rapportés chez des nouveau-nés de mères traitées avec du valproate pendant la grossesse.
· Un syndrome de sevrage (en particulier agitation, irritabilité, hyperexcitabilité, nervosité, hyperkinésie, troubles du tonus, tremblements, convulsions et troubles de l’alimentation) peut survenir chez les nouveau-nés de mères traitées avec du valproate pendant le troisième trimestre de la grossesse.
Suivi post-natal/chez l’enfant
En cas d’exposition pendant la grossesse, un suivi rapproché du développement neurocomportemental de l’enfant est à instaurer et une prise en charge adaptée doit être mise en place au plus tôt en cas de nécessité.
Adolescents et hommes en âge de procréer :
Risque potentiel de troubles neuro-développementaux chez les enfants dont le père a été traité par le valproate dans les 3 mois précédant la conception
Une étude observationnelle rétrospective conduite dans 3 pays nordiques suggère une augmentation du risque de troubles neuro-développementaux (TND) chez les enfants (de 0 à 11 ans) nés de père traité par valproate en monothérapie dans les 3 mois précédant la conception comparativement à ceux dont le père était traité par lamotrigine ou lévétiracétam en monothérapie, avec un Hazard Ratio (HR) ajusté de 1,50 (IC à 95 % : 1,09-2,07). Le risque cumulé ajusté de TND était compris entre 4,0 % et 5,6 % dans le groupe valproate contre 2,3 % à 3,2 % dans le groupe composite lamotrigine/ lévétiracétam. Le nombre de patients inclus dans l'étude n'était pas suffisant pour étudier les associations avec des sous-types spécifiques de TND.
Les limites de l’étude incluaient un facteur potentiel de confusion lié à l’indication et des différences dans la durée de suivi des groupes d’exposition. La durée moyenne de suivi des enfants du groupe valproate était comprise entre 5 et 9,2 ans contre 4,8 et 6,6 ans pour les enfants du groupe lamotrigine/ lévétiracétam. Un risque accru de TND chez les enfants de pères traités par valproate dans les 3 mois précédant la conception est possible, cependant le lien de causalité avec le valproate n’est pas confirmé. En outre, l’étude n’a pas évalué le risque de TND chez les enfants nés d’hommes arrêtant le valproate pendant plus de 3 mois avant la conception (c’est-à-dire permettant une nouvelle spermatogenèse sans exposition au valproate).
Par mesure de précaution, le prescripteur doit informer les adolescents et hommes en âge de procréer de ce risque potentiel et discuter avec eux :
· de la nécessité de mettre en place des mesures contraceptives efficaces pendant le traitement par valproate et au moins trois mois après l’arrêt de celui-ci, y compris pour leur partenaire (voir rubrique 4.4),
· de ne pas faire de don de sperme pendant le traitement par valproate et au moins trois mois après l’arrêt du valproate.
Le traitement par valproate chez les hommes et adolescents en âge de procréer doit être régulièrement réévalué par le prescripteur afin de déterminer si le valproate reste le traitement le plus approprié. Pour les patients de sexe masculin prévoyant de concevoir un enfant, des alternatives thérapeutiques doivent être envisagées et discutées avec les patients. Les situations individuelles doivent être évaluées dans chaque cas. Il est recommandé de demander conseil à un médecin spécialisé dans la prise en charge de l’épilepsie si nécessaire.
Une attestation d’information partagée doit être utilisée, dûment complétée et signée au moment de l’instauration du traitement par le médecin spécialiste et lors de chaque réévaluation annuelle du traitement par valproate par tout médecin et le patient (ou son représentant légal).
Le valproate est excrété dans le lait maternel à une concentration comprise entre 1 % et 10 % des niveaux sériques maternels. Des troubles hématologiques ont été observés chez des nouveau-nés/nourrissons allaités par des femmes sous traitement (voir rubrique 4.8).
La décision d’interrompre l'allaitement ou de suspendre le traitement par DEPAKINE doit tenir compte du bénéfice de l'allaitement pour l'enfant et du bénéfice du traitement pour la femme.
Fertilité
Des cas d'aménorrhée, d’ovaires polykystiques et d'augmentation des taux de testostérone ont été rapportés chez des femmes traitées avec du valproate (voir rubrique 4.8).
Chez l'homme, l’administration du valproate peut également nuire à la fertilité (diminution de la mobilité des spermatozoïdes en particulier) (voir rubrique 4.8). Dans quelques cas, ces troubles de la fertilité sont réversibles après au moins 3 mois d'arrêt du traitement. Dans un nombre limité de cas, il a été rapporté qu’une réduction importante de la posologie est susceptible d’améliorer la fertilité. Cependant, dans d’autres cas, la réversibilité de ces troubles de la fertilité masculine n’est pas connue.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Classification des fréquences attendues :
Très fréquent (≥ 10 %) ; Fréquent (≥1 % - <10 %) ; Peu fréquent (≥ 0,1 % - <1 %) ; Rare (≥ 0,01 % - < 0,1 %) ; Très rare (<0,01 %) ; Indéterminée (ne peut être estimée d’après les données disponibles).
Affections congénitales, familiales et génétiques
· Malformations congénitales, troubles neuro-développementaux (voir rubriques 4.4 et 4.6).
Affections hématologiques et du système lymphatique
· Fréquent : anémie, thrombopénie.
Des cas de thrombopénie dose-dépendante, généralement de découverte systématique et sans retentissement clinique, ont été décrits.
En cas de thrombopénie asymptomatique, si le taux de plaquettes et si le contrôle de la maladie le permettent, la seule diminution de posologie de ce médicament permet le plus souvent la régression de cette thrombopénie.
· Peu fréquent : leucopénie, pancytopénie.
· Rare : aplasie médullaire globale ou aplasie pure de la lignée rouge, agranulocytose, anémie macrocytaire, macrocytose.
Investigations
· Fréquent : prises de poids*.
· Rare : diminution d’au moins un facteur de coagulation, tests de coagulation anormaux (tel que allongement du temps de prothrombine, allongement du temps de céphaline activé, allongement du temps de thrombine, augmentation de l’INR) (voir rubriques 4.4 et 4.6), déficit en vitamine B8 (biotine)/déficit en biotinidase.
Fréquence indéterminée : anomalie acquise de Pelger-Huët**. *Les prises de poids étant un facteur de risque de survenue du syndrome des ovaires polykystiques, le poids des patientes doit faire l’objet d’une surveillance attentive (voir rubrique 4.4).
**Une anomalie acquise de Pelger-Huët a été rapportée, avec et sans syndrome myélodysplasique.
Affections du système nerveux
· Très fréquent : tremblements
· Fréquent : troubles extrapyramidaux**, stupeur*, sédation, convulsion*, troubles de la mémoire, céphalées, nystagmus, sensations nauséeuses ou vertigineuses
· Peu fréquent : coma*, encéphalopathie*, léthargie*, syndromes parkinsoniens réversibles**, ataxie, paresthésie
· Rare : diplopie, troubles cognitifs d’installation insidieuse et progressive (pouvant réaliser un tableau complet de syndrome démentiel) réversibles quelques semaines à quelques mois après l’arrêt du traitement**
*Des cas d'états stuporeux ou de léthargie aboutissant parfois à un coma transitoire (encéphalopathie) sous valproate, ont été observés, régressant à l'arrêt du traitement ou à la diminution des doses. Ces états surviennent le plus souvent lors de polythérapies (phénobarbital ou topiramate en particulier) ou d'augmentation brusque des doses de valproate.
**Ces symptômes peuvent être associés à une imagerie d’atrophie cérébrale.
Affections de l’oreille et du labyrinthe
· Fréquent : pertes d’audition.
Affections respiratoires, thoraciques et médiastinales
· Peu fréquent : épanchement pleural (éosinophilique).
Affections gastro-intestinales
· Très fréquent : nausées.
· Fréquent : vomissements, troubles gingivaux (principalement hyperplasie gingivale), stomatite, douleurs épigastriques, diarrhées qui peuvent survenir chez certains patients en début de traitement, mais qui cèdent en général au bout de quelques jours sans interruption du traitement.
· Peu fréquent : pancréatite dont l’évolution peut être fatale et qui nécessite un arrêt précoce du traitement (voir rubrique 4.4).
Affections du rein et des voies urinaires
· Fréquent : incontinence urinaire.
· Peu fréquent : insuffisance rénale.
· Rare : énurésie, néphrite tubulo-interstitielle, syndrome de Fanconi.
Affections de la peau et du tissu sous-cutané
· Fréquent : chute des cheveux passagère et/ou dose-dépendante, troubles de l’ongle et du lit de l’ongle.
· Peu fréquent : angioœdème, réactions cutanées, troubles capillaires (tels que texture anormale des cheveux, changements de la couleur des cheveux, pousse anormale des cheveux)
· Rare : syndrome de Lyell, syndrome de Stevens-Johnson, érythème polymorphe, syndrome DRESS (Drug Rash with Eosinophilia and Systemic Symptoms) ou syndrome d’hypersensibilité médicamenteuse.
· Fréquence indéterminée : hyperpigmentation.
Affections endocriniennes
· Peu fréquent : syndrome de sécrétion inappropriée de l'hormone anti-diurétique (SIADH), hyperandrogénie (hirsutisme, virilisme, acné, alopécie de type androgénique, et/ou augmentation du taux d’hormones androgènes).
· Rare : hypothyroïdie (voir rubrique 4.6).
Troubles du métabolisme et de la nutrition
· Fréquent : hyponatrémie.
· Rare : hyperammoniémie* (voir rubrique 4.4), obésité.
*Une hyperammoniémie isolée et modérée sans modification des tests biologiques hépatiques peut être observée, surtout en cas de polythérapie, et ne doit pas faire interrompre le traitement.
Toutefois, des cas d'hyperammoniémie avec symptômes neurologiques (pouvant aller jusqu'au coma) ont aussi été rapportés, nécessitant alors des investigations complémentaires (voir les rubriques 4.3 et 4.4 : « Troubles du cycle de l’urée et risque d’hyperammoniémie et Patients à risque d’hypocarnitinémie »).
· Fréquence indéterminée : hypocarnitinémie (voir rubriques 4.3 et 4.4).
Tumeurs bénignes, malignes, et non précisées (incl. kystes et polypes)
· Rare : syndrome myélodysplasique
Affections vasculaires
· Fréquent : hémorragie (voir rubriques 4.4 et 4.8)
· Peu fréquent : vascularite cutanée, essentiellement vascularite leucocytoclasique.
Troubles généraux et anomalies au site d’administration
· Peu fréquent : hypothermie, œdème périphérique non sévère.
Affections hépatobiliaires
· Fréquent : hépatopathies (voir rubrique 4.4).
Affections des organes de reproduction et du sein
· Fréquent : irrégularités menstruelles
· Peu fréquent : aménorrhées
· Rare : troubles de la fertilité masculine, (voir rubrique 4.6), ovaires polykystiques.
Affections musculo-squelettiques et systémiques
· Peu fréquent : diminution de la densité minérale osseuse, ostéopénie, ostéoporose et fractures chez des patients traités au long cours par DEPAKINE. Le mode d’action de DEPAKINE sur le métabolisme osseux n’est pas connu.
· Rare : lupus érythémateux aigu disséminé (voir rubrique 4.4), rhabdomyolyse (voir rubrique 4.4).
Affections psychiatriques
· Fréquent : état confusionnel, hallucinations, agressivité*, agitation*, troubles de l’attention*.
· Rare : comportement anormal*, hyperactivité psychomotrice*, difficultés d’apprentissage*.
*Ces effets sont observés essentiellement dans la population pédiatrique.
Population pédiatrique
Le profil de sécurité du valproate dans la population pédiatrique est comparable à celui des adultes, mais certains effets indésirables sont plus graves ou sont principalement observés dans la population pédiatrique. Il existe un risque particulier d'atteinte hépatique sévère chez les nourrissons et les jeunes enfants, en particulier avant l'âge de 3 ans. Les jeunes enfants ont également un risque particulier de pancréatite. Ces risques diminuent avec l'âge (voir rubrique 4.4). Les troubles psychiatriques tels que l'agressivité, l'agitation, les troubles de l'attention, les comportements anormaux, l'hyperactivité psychomotrice et les troubles d'apprentissage sont principalement observés dans la population pédiatrique.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament.
Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Le tableau de l'intoxication aiguë massive comporte habituellement un coma calme, plus ou moins profond, avec hypotonie musculaire, hyporéflexie, myosis, diminution de l'autonomie respiratoire, acidose métabolique, hypotension et collapsus/choc cardio-vasculaire.
Quelques cas d'hypertension intracrânienne liée à un œdème cérébral ont été décrits.
Les mesures à entreprendre en milieu hospitalier sont : évacuation gastrique si indiquée, maintien d'une diurèse efficace, surveillance cardiorespiratoire. Dans les cas très graves, on pratiquera éventuellement une épuration extra- rénale.
Le pronostic de ces intoxications est généralement favorable, cependant quelques décès ont été rapportés.
La présence de sodium dans les formulations contenant du valproate peut entraîner une hypernatrémie en cas de surdosage.
Traitement
En cas de surdosage en valproate entraînant une hyperammoniémie, de la carnitine peut être administrée par voie IV pour tenter de normaliser les taux d’ammonium.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : ANTIEPILEPTIQUE, code ATC : N03AG01.
Le valproate exerce ses effets pharmacologiques essentiellement au niveau du système nerveux central.
Ces propriétés anticonvulsivantes s'exercent contre des types très variés de crises convulsives chez l'animal et d'épilepsies chez l'homme.
Les études expérimentales et cliniques du valproate suggèrent deux types d'action anti-convulsivantes.
Le premier est un effet pharmacologique direct en relation avec les concentrations en valproate du plasma et du cerveau.
Le second est apparemment indirect et vraisemblablement en relation avec des métabolites du valproate persistant dans le cerveau ou avec des modifications des neurotransmetteurs ou avec des effets membranaires directs. L'hypothèse la plus généralement admise est l'hypothèse de l'acide gamma- aminobutyrique (GABA) dont le taux augmente après administration de valproate.
Le valproate diminue la durée des phases intermédiaires de sommeil avec une augmentation concomitante de sommeil lent.
5.2. Propriétés pharmacocinétiques
Les différentes études pharmacocinétiques effectuées pour le valproate, ont montré que :
· La biodisponibilité sanguine du valproate après administration orale est proche de 100 %.
· Le volume de distribution est limité essentiellement au sang et aux liquides extra-cellulaires à échange rapide. Le valproate diffuse dans le L.C.R. et dans le cerveau.
· Passage de la barrière placentaire (voir rubrique 4.6) :
Le valproate traverse la barrière placentaire chez l’animal et chez l’Homme :
o chez l’animal, le valproate traverse le placenta, de manière similaire à l’Homme,
o chez l’Homme, plusieurs publications ont évalué la concentration du valproate dans le cordon ombilical des nouveau-nés à l’accouchement. La concentration sérique du valproate dans le cordon ombilical, représentant la concentration sérique du valproate chez les fœtus, était similaire ou légèrement supérieure à celle des mères.
· La demi-vie est de 15 à 17 heures.
· L'efficacité thérapeutique nécessite habituellement une concentration sérique minimale de 40-50 mg/l, avec une large fourchette comprise entre 40 et 100 mg/l. Si des taux plasmatiques supérieurs s'avèrent nécessaires, les bénéfices attendus doivent être pesés par rapport au risque de survenue d'effets indésirables en particulier dose-dépendants. Toutefois, des taux se maintenant au-delà de 150 mg/l nécessitent une réduction de la posologie.
· La concentration plasmatique d'équilibre est atteinte en 3 à 4 jours.
· La fixation protéique du valproate est très importante. Elle est dose-dépendante et saturable.
· La voie majeure du métabolisme du valproate est la glucuro-conjugaison (environ 40 %), principalement via l’UGT1A6, l’UGT1A9 et l’UGT2B7.
· L'excrétion du valproate est essentiellement urinaire après métabolisation par glucuro-conjugaison et bêta- oxydation.
· La molécule de valproate est dialysable, mais l'hémodialyse ne touche que la fraction libre de valproate sanguin (environ 10 %).
· Le valproate n'est pas inducteur des enzymes impliquées dans le système métabolique du cytochrome P 450 : contrairement à la plupart des autres antiépileptiques, il n'accélère pas de ce fait sa propre dégradation, ni celle d'autres substances telles que les œstroprogestatifs et les antivitamines K.
Population pédiatrique
A partir de l'âge de 10 ans, les enfants et les adolescents ont des clairances de valproate similaires à celles rapportées chez les adultes. Chez les patients pédiatriques de moins de 10 ans, la clairance systémique du valproate varie avec l'âge.
Chez les nouveau-nés et les nourrissons jusqu'à l'âge de 2 mois, la clairance du valproate est diminuée par rapport à celle des adultes et elle est la plus faible immédiatement après la naissance. Dans une revue de la littérature scientifique, la demi-vie du valproate chez les nourrissons de moins de deux mois a montré une variabilité considérable allant de 1 à 67 heures.
Chez les enfants âgés de 2 à 10 ans, la clairance du valproate est 50 % plus élevée que chez les adultes.
5.3. Données de sécurité préclinique
In vitro, le valproate n'a été mutagène ni dans les bactéries, ni dans les essais sur lymphome de souris et n'a pas induit de réparation de l'ADN dans les cultures primaires d'hépatocytes de rat. Cependant, in vivo, des résultats contradictoires ont été obtenus à des doses tératogènes selon la voie d'administration. Après administration orale, voie prédominante chez l'Homme, le valproate n'a induit ni d'aberrations chromosomiques dans la moelle osseuse de rat, ni d'effets létaux majeurs chez la souris. Une injection intrapéritonéale de valproate a augmenté les ruptures de brins d'ADN et les aberrations chromosomiques chez les rongeurs. De plus, une augmentation des échanges de chromatides sœurs chez les patients épileptiques exposés au valproate a été rapportée dans des études publiées comparée aux sujets sains non traités. Cependant, des résultats contradictoires ont été obtenus en comparant les données des patients épileptiques traités par valproate avec celles des patients épileptiques non traités. La pertinence clinique de ces conclusions sur l’ADN/chromosome n’est pas connue.
Les données non cliniques issues des études conventionnelles de carcinogénicité ne révèlent aucun risque particulier pour l'Homme.
Toxicité sur la reproduction
Le valproate a induit des effets tératogènes (malformations de plusieurs systèmes d'organes) chez la souris, le rat et le lapin.
Chez les souris, les rats et les lapins, l'exposition in utero au valproate a induit une diminution dose-dépendante du poids fœtal, un retard de croissance intra-utérin et une réduction de la longueur vertex-coccyx par rapport aux animaux non exposés.
Des anomalies comportementales ont été rapportées chez des descendants de première génération de souris et de rats après exposition in utero. Certains changements de comportement ont également été observés dans la deuxième génération et ceux-ci étaient moins prononcés dans la troisième génération de souris après exposition aiguë in utero de la première génération à des doses tératogènes de valproate. Les mécanismes sous-jacents et la pertinence clinique de ces résultats ne sont pas connus.
Dans les études de toxicité à doses répétées, une dégénérescence/atrophie des testicules, une spermatogenèse anormale et une diminution du poids des testicules ont été rapportées chez des rats et des chiens adultes après administration orale de doses de 400 mg/kg/jour et 150 mg/kg/jour, respectivement. Les doses sans effet nocif observable (DSENO) associées aux effets testiculaires sont de 270 mg/kg/jour chez le rat adulte et de 90 mg/kg/jour chez le chien adulte.
Sur la base des extrapolations d’ASC (Aire Sous Courbe) chez le rat et le chien, il pourrait ne pas y avoir de marge de sécurité pour l’humain.
Chez les rats juvéniles, la diminution du poids des testicules n’a été observée qu’à des doses dépassant la dose maximale tolérée (à partir de 240 mg/kg/jour par voie intrapéritonéale ou intraveineuse) et sans modification histopathologique associée. Aucun effet sur les organes reproducteurs mâles n’a été observé aux doses tolérées (jusqu’à 90 mg/kg/jour). Au vu de ces données, les animaux juvéniles n’ont pas été jugés plus susceptibles que les adultes de présenter des troubles testiculaires. La pertinence clinique de ces résultats sur les testicules pour la population pédiatrique demeure inconnue.
Lors d’une étude sur la fertilité chez les rats, l’administration de valproate à des doses allant jusqu’à 350 mg/kg/jour n’a pas altéré les performances de reproduction chez les mâles. Cependant, les troubles de la fertilité masculine ont été identifiés comme un effet indésirable chez l’Homme (voir les rubriques 4.6 et 4.8).
Urée, solution d'hydroxyde de sodium à 30 %, eau purifiée.
3 ans.
6.4. Précautions particulières de conservation
Pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Flacon de 40 ml en verre brun avec bouchon en polyéthylène.
6.6. Précautions particulières d’élimination et de manipulation
Mode d’administration :
|
|
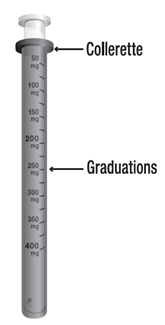
La solution buvable sera absorbée après dilution dans un peu de boisson non gazeuse. Le flacon de solution buvable est accompagné d’une seringue pour administration orale (piston rose) uniquement réservée à l’utilisation de ce médicament. Cette seringue pour administration orale est graduée de 50 mg à 400 mg, tous les 50 mg avec des graduations secondaires de 25 mg, et elle porte la mention « DEPAKINE 200 mg/ml, solution buvable ». Les traits de graduation indiquent la dose exprimée en milligrammes.
|

|
Ouverture du flacon Pour ouvrir le flacon, tourner le bouchon sécurité-enfant en appuyant sur le dessus.
|
|
Utilisation de la seringue pour administration orale
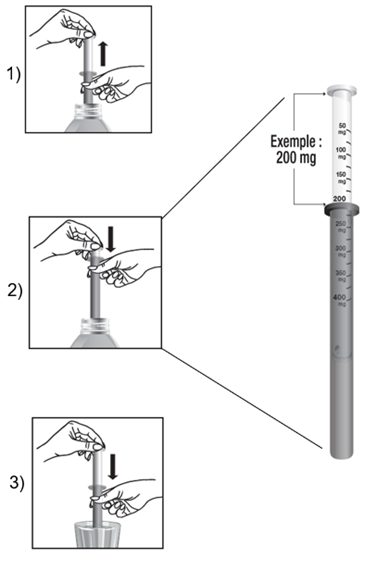
· La dose à administrer est obtenue en tirant le piston jusqu’à la graduation correspondant à la quantité en milligrammes (mg) prescrite.
· La dose est lue au niveau de la collerette de la seringue pour administration orale.
Exemple pour une dose de 200 mg :
|
1) Remplir la seringue pour administration orale en tirant le piston.
2) Ajuster la dose à 200 mg.
3) Vider le contenu de la seringue pour administration orale dans un verre et le diluer dans un peu de boisson non gazeuse.
|
|
Après utilisation, refermer le flacon, bien rincer à l’eau et sécher la seringue pour administration orale.
Puis ranger immédiatement la seringue pour administration orale au sein de sa boîte dans un endroit inaccessible aux enfants. Ne jamais séparer la seringue pour administration orale des autres éléments de conditionnement du médicament (boîte, notice).
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
82, AVENUE RASPAIL
94250 GENTILLY
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 930 0 3 : 40 ml en flacon (verre brun) avec seringue pour administration orale (polyéthylène).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II
Enfants et adolescents de sexe féminin, femmes susceptibles de procréer et femmes enceintes : Prescription initiale annuelle réservée aux spécialistes en neurologie ou en pédiatrie. Renouvellement non restreint.
Adolescents de sexe masculin et hommes susceptibles de procréer : Prescription initiale réservée aux spécialistes en neurologie ou en pédiatrie. Renouvellement non restreint.
Médicament nécessitant une surveillance particulière pendant le traitement : la prescription nécessite la signature annuelle par le médecin et la patiente ou le patient de l’attestation d’information partagée. La délivrance nécessite la présentation de l’attestation annuelle co-signée.
: Ce médicament fait l'objet d'une surveillance renforcée. Pour plus d'informations, cliquez ici
Informations importantes
Les informations importantes disponibles pour ce médicament sont les suivantes :
- Valproate et dérivés : réduire le risque de troubles neurodéveloppementaux chez les enfants dont le père a été traité avant la conception
- Exposition paternelle au valproate pendant la période de conception : nouvelle étude en faveur d'un risque accru de troubles neuro-développementaux chez l'enfant
- Antiépileptiques pendant la grossesse entre 2013 et 2021 : une étude du GIS EPI-PHARE met en évidence une baisse importante de l'exposition au valproate et dérivés, mais moindre pour le topiramate et la carbamazépine
- Valproate et risques pour l'enfant à naître : les conditions de prescription et de délivrance évoluent pour les adolescents et les hommes susceptibles d'avoir des enfants
- Retour d'information sur le PRAC de janvier 2024 (8 - 11 janvier)
- Antiépileptiques et grossesse : mieux connaître les risques pour l'enfant à naître
- Valproate et dérivés : risque potentiel de troubles neurodéveloppementaux chez les enfants dont le père a été traité dans les 3 mois qui précèdent la conception
- Evaluation européenne du risque potentiel de troubles neurodéveloppementaux chez les enfants dont le père a été traité par valproate dans les mois précédant la conception
- Valproate et dérivés : mise à jour des informations sur le risque
ANSM - Mis à jour le : 30/10/2025
DEPAKINE 200 mg/ml, solution buvable
Valproate de sodium
[Code QR (Quick Response) : l’emballage et la notice doivent comporter un code QR, dont l’emplacement sera choisi en prenant en compte la lisibilité générale.]
MISE EN GARDE
DEPAKINE PEUT NUIRE GRAVEMENT A L’ENFANT A NAITRE S’IL EST PRIS PENDANT LA GROSSESSE.
Les enfants exposés in utero au valproate présentent un risque élevé de troubles graves du développement (intellectuel et moteur) et du comportement (jusqu’à 30 à 40 % des cas) et/ou de malformations (environ 11 % des cas).
Si vous êtes une fille, une adolescente, une femme en âge d’avoir des enfants :
· votre médecin spécialiste ne pourra pas vous prescrire de valproate, sauf en cas d’inefficacité ou d’intolérance aux autres traitements ;
· si aucun autre traitement n’est possible, le valproate vous sera prescrit et dispensé sous des conditions très strictes d’un programme de prévention de la grossesse ayant pour but d’éviter toute grossesse.
Si du valproate vous a été prescrit et que vous êtes une femme en âge d’avoir des enfants, vous devez notamment :
· utiliser au moins une méthode de contraception efficace, sans interruption, pendant toute la durée de votre traitement par DEPAKINE. Votre médecin discutera de cela avec vous mais vous devez également suivre les conseils donnés à la rubrique 2 de cette notice ;
· prendre rendez-vous en urgence avec votre médecin spécialiste si vous envisagez une grossesse ou pensez être enceinte ;
· ne pas arrêter de prendre DEPAKINE sans que votre médecin ne vous l’ait demandé ; cela pourrait aggraver votre maladie.
Assurez-vous d’avoir lu et compris la brochure d’information patiente et d’avoir signé l’attestation d’information partagée qui vous sera remise par votre médecin spécialiste expérimenté dans la prise en charge de l’épilepsie.
Demandez conseil à votre médecin ou pharmacien.
Si vous êtes un homme en âge d’avoir des enfants et que du valproate vous a été prescrit, reportez-vous au paragraphe « Grossesse, allaitement et fertilité – Informations importantes à l'attention des adolescents et hommes en âge d’avoir des enfants ».
Assurez-vous d’avoir lu et compris la brochure d’information patient et d’avoir signé l’attestation d’information partagée qui vous sera remise par votre médecin spécialiste à l’instauration du traitement puis par votre médecin chaque année.
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
Que contient cette notice ?
1. Qu'est-ce que DEPAKINE 200 mg/ml, solution buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre DEPAKINE 200 mg/ml, solution buvable ?
3. Comment prendre DEPAKINE 200 mg/ml, solution buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver DEPAKINE 200 mg/ml, solution buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE DEPAKINE 200 mg/ml, solution buvable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique ANTIEPILEPTIQUE - code ATC N03AG01
DEPAKINE appartient à une famille de médicaments appelée antiépileptiques.
Ce médicament est utilisé pour le traitement des différentes formes d’épilepsie chez l’adulte et l’enfant. Chez l’enfant, il est également utilisé pour le traitement préventif des convulsions liées à la fièvre.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE DEPAKINE 200 mg/ml, solution buvable ?
Ne prenez jamais DEPAKINE 200 mg/ml, solution buvable
· si vous êtes enceinte, sauf si aucun autre traitement de l’épilepsie n’est efficace pour vous (voir paragraphe « Grossesse, allaitement et fertilité - Conseils importants à l’attention des femmes »),
· si vous êtes une femme en âge d’avoir des enfants, sauf si aucun autre traitement de l’épilepsie n’est efficace pour vous et que vous êtes capable de respecter toutes les mesures du plan de prévention pour éviter une grossesse (voir paragraphe « Grossesse, allaitement et fertilité - Conseils importants à l’attention des femmes »),
· si vous êtes allergique au valproate de sodium ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6,
· si vous êtes allergique à un médicament de la même famille que le valproate (divalproate, valpromide),
· si vous avez une maladie du foie (hépatite aiguë ou chronique),
· si vous ou un membre de votre famille avez déjà eu une hépatite grave notamment liée à la prise d’un médicament,
· si vous souffrez d’une porphyrie hépatique (maladie héréditaire du foie),
· si vous avez un problème génétique causant un trouble mitochondrial (par ex. le syndrome d’Alpers-Huttenlocher),
· si vous avez un trouble du métabolisme connu, par exemple un trouble du cycle de l’urée (voir paragraphe « Avertissements et précautions »),
· si vous présentez une déficience en carnitine (une maladie métabolique très rare) non traitée,
· si vous prenez en même temps du millepertuis (plante servant à traiter la dépression).
Avertissements et précautions
|
Ce médicament peut très rarement provoquer une atteinte du foie (hépatite) ou du pancréas (pancréatite) pouvant être grave et mettre votre vie en danger. Votre médecin vous prescrira des examens du sang pour surveiller régulièrement le fonctionnement de votre foie, notamment au cours des 6 premiers mois de traitement. Prévenez immédiatement votre médecin en cas d’apparition des signes suivants : · Des réactions cutanées graves, dont le syndrome de Stevens-Johnson, la nécrolyse épidermique toxique, le syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), un érythème polymorphe et un angioœdème, ont été rapportées en lien avec un traitement par valproate. Consultez immédiatement un médecin si vous remarquez l’un des symptômes liés à ces réactions cutanées graves décrites à la rubrique 4 ; · fatigue soudaine, perte d’appétit, abattement, somnolence, gonflement des jambes, malaise général, · vomissements répétés, nausées, douleurs dans le ventre et à l’estomac, coloration jaune de la peau ou des yeux (jaunisse), · réapparition des crises alors que vous suivez correctement votre traitement, · fièvre et difficulté à respirer. |
· Le risque d’atteinte du foie est accru si DEPAKINE est administré à des enfants de moins de 3 ans, des personnes prenant d’autres médicaments antiépileptiques en même temps ou des personnes présentant une autre maladie neurologique ou métabolique et des formes sévères d’épilepsie.
· Si lors du traitement par DEPAKINE, vous ou votre enfant présentez des problèmes d’équilibre et de coordination, une sensation de léthargie ou de moins bonne vigilance ou encore des vomissements, informez immédiatement votre médecin. Cela peut être dû à une quantité accrue d’ammonium dans le sang.
Avant de prendre ce médicament, prévenez votre médecin :
· Si vous avez une maladie des reins (insuffisance rénale).
· Si vous avez un lupus érythémateux disséminé (maladie rare).
· Si vous savez ou si votre médecin soupçonne qu'il existe un problème génétique causé par une maladie mitochondriale dans votre famille, en raison d'un risque d'atteinte de votre foie.
· Si vous êtes suspecté de souffrir de troubles métaboliques, notamment de troubles héréditaires par déficit enzymatique de type « trouble du cycle de l'urée » en raison d’un risque d'augmentation de la quantité d'ammonium dans le sang.
· Si vous souffrez d’une maladie rare (maladie métabolique héréditaire) appelée « déficit en carnitine palmitoyl transférase de type II », car vous présentez un risque accru de troubles musculaires graves (rhabdomyolyse).
· Si vous avez une carence alimentaire en carnitine, présente dans la viande et les produits laitiers, en particulier chez les enfants de moins de 10 ans.
· Si vous avez une carence en carnitine et que vous prenez de la carnitine.
· Si vous devez subir une intervention chirurgicale, vous devez prévenir le personnel médical que vous prenez ce médicament.
· En début de traitement, le médecin s’assurera que vous n'êtes pas enceinte et que vous avez un moyen de contraception (voir paragraphe « Grossesse »).
· Comme avec les autres antiépileptiques, suite à la prise de ce médicament, les crises peuvent s’aggraver, survenir plus fréquemment ou des crises de type différent peuvent apparaître. Dans ce cas, consultez immédiatement votre médecin.
· Ce médicament peut entraîner une prise de poids. Votre médecin vous conseillera de prendre certaines mesures diététiques et surveillera votre poids.
· Des pensées autodestructrices ou suicidaires ont également été observées chez un petit nombre de personnes traitées par des antiépileptiques tels que DÉPAKINE. Si vous avez ce type de pensées, contactez immédiatement votre médecin.
· En cas d’apparition de symptômes tels que des tremblements, rigidité des membres et difficultés pour marcher (troubles extrapyramidaux) ou des troubles de la mémoire et des capacités mentales, informez votre médecin. Une pathologie sous-jacente ou la responsabilité de DEPAKINE doivent être recherchées. Un arrêt du traitement pourrait être nécessaire.
Prévenez votre médecin si votre enfant prend un autre traitement antiépileptique ou souffre d’une autre maladie neurologique ou métabolique et de formes sévères d’épilepsie.
Autres médicaments et DEPAKINE 200 mg/ml, solution buvable :
Certains médicaments peuvent influencer les effets du valproate et inversement.
Vous ne devez jamais prendre ce médicament si vous prenez le médicament suivant :
· du millepertuis (médicament à base de plante servant à traiter la dépression).
Vous ne devez pas prendre ce médicament, sauf avis contraire de votre médecin ou votre pharmacien, si vous prenez, avez récemment pris ou pourriez prendre les médicaments suivants :
· de la lamotrigine (autre médicament utilisé pour traiter des crises d'épilepsie),
· des pénems (carbapénèmes) - (antibiotiques utilisés pour traiter les infections bactériennes).
Informez votre médecin si vous prenez :
· des médicaments contenant de l’acétazolamide (médicaments utilisés pour diminuer la pression au niveau de l’œil ou le taux de gaz carbonique dans le sang),
· des médicaments antibiotiques (médicaments contenant de l’aztréonam ou de la rifampicine),
· d’autres médicaments antiépileptiques (médicaments contenant de la carbamazépine, du felbamate, de la phénytoïne, de la fosphénytoïne, de la primidone, du phénobarbital, du rufinamide, du topiramate ou du zonisamide),
· de la nimodipine DEPAKINE peut augmenter les effets de la nimodipine (médicament utilisé pour prévenir les complications qui peuvent survenir après un saignement au niveau du cerveau),
· des médicaments contenant des œstrogènes (y compris certaines pilules contraceptives),
· du propofol (médicament anesthésique),
· des médicaments contenant de la zidovudine (médicaments utilisés pour traiter l’infection par le VIH (Virus de l’Immunodéficience Humaine)),
· des médicaments contenant du lithium (médicaments utilisés pour traiter des troubles de l’humeur),
· de la clozapine (pour traiter les problèmes de santé mentale),
· des médicaments contenant du métamizole (médicaments utilisés pour traiter la douleur et la fièvre),
· du méthotrexate (utilisé pour traiter des cancers et des maladies inflammatoires),
· des dérivés salicylés (dont l’aspirine),
· du cannabidiol (utilisé pour traiter l’épilepsie et d’autres maladies),
· certains anti-infectieux contenant du pivalate (par exemple : pivampicilline, adéfovir, dipivoxil).
Chez l’enfant spécifiquement de moins de 3 ans, vous devez éviter de donner des médicaments contenant des dérivés salicylés (dont l’aspirine) au cours du traitement.
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
DEPAKINE 200 mg/ml, solution buvable avec des aliments, boissons et de l’alcool
La prise de boissons alcoolisées est déconseillée pendant la durée du traitement par DEPAKINE.
Grossesse, allaitement et fertilité
Grossesse
Conseils importants à l’attention des femmes :
Le valproate est dangereux pour l’enfant à naître s’il est pris pendant la grossesse. Par conséquent :
· Si vous êtes une fille, une adolescente, une femme en âge d’avoir des enfants, votre médecin spécialiste ne pourra pas vous prescrire de valproate, sauf en cas d’inefficacité ou d’intolérance aux autres traitements. Si aucun autre traitement n’est possible, le valproate vous sera prescrit et dispensé sous des conditions très strictes, décrites ci-dessous.
· Assurez-vous d’avoir lu la brochure d’information patiente remise par votre médecin spécialiste. Votre médecin discutera avec vous de l’attestation d’information partagée et vous demandera de la signer et de la conserver. Vous devrez la présenter au pharmacien lors de chaque délivrance, ainsi que l’ordonnance du spécialiste. Ce document atteste que l’on vous a bien expliqué les risques et que vous acceptez de respecter les conditions ci-dessous. Votre pharmacien vous remettra par ailleurs une carte patiente qui vous rappelle les risques liés à la prise de valproate pendant la grossesse.
Vous ne devez pas prendre DEPAKINE :
· Si vous êtes enceinte, sauf si aucun autre traitement de l’épilepsie n’est efficace pour vous.
· Si vous êtes une femme en âge d’avoir des enfants, sauf si aucun autre traitement de l’épilepsie n’est efficace pour vous et que vous êtes capable de respecter toutes les mesures du plan de prévention pour éviter une grossesse.
Risques liés à la prise de valproate pendant la grossesse
· Adressez-vous immédiatement à votre médecin spécialiste si vous prévoyez d’avoir un enfant, si vous êtes enceinte ou pensez l’être.
· Le valproate expose à un risque pour l’enfant à naître s’il est pris pendant la grossesse. Plus la dose est élevée, plus les risques sont importants toutefois, toutes les doses exposent à un risque, y compris lorsque le valproate est utilisé en association avec d’autres médicaments pour traiter l’épilepsie.
· S’il est pris par une femme enceinte, le valproate peut provoquer de graves malformations congénitales et peut nuire au développement (intellectuel, moteur, comportemental) de l’enfant pendant sa croissance.
· Les malformations congénitales les plus fréquemment rapportées incluent le spina bifida (malformation osseuse de la colonne vertébrale), des malformations de la face, de la lèvre supérieure et du palais, du crâne, du cœur, des reins, des voies urinaires et des organes génitaux ainsi que des atteintes des membres et de multiples malformations associées affectant plusieurs organes et parties du corps. Les malformations congénitales peuvent entraîner des handicaps qui peuvent être sévères.
· Des problèmes auditifs ou une surdité ont été rapportés chez les enfants exposés au valproate pendant la grossesse.
· Si vous prenez du valproate pendant la grossesse, vous avez un risque plus élevé que les autres femmes d’avoir un enfant atteint de malformations nécessitant un traitement médical. Le valproate étant utilisé depuis de nombreuses années, il est établi que près de 11 bébés sur 100 nés de mères sous valproate présentent des malformations, contre 2 à 3 bébés sur 100 dans la population générale.
· On estime que jusqu'à 30 à 40 % des enfants d’âge préscolaire dont les mères ont pris du valproate pendant la grossesse présentent des problèmes de développement dans leur petite enfance. Les enfants concernés marchent et/ou parlent plus tardivement, et/ou ont des capacités intellectuelles plus faibles que les autres enfants, et/ou ont des difficultés de langage, et/ou de mémoire.
· Les troubles du spectre autistique sont plus souvent diagnostiqués chez les enfants exposés au valproate pendant la grossesse.
· Des données indiquent que les enfants exposés au valproate pendant la grossesse ont un risque accru de développer le trouble de déficit de l’attention/hyperactivité (TDAH).
· Si vous prenez du valproate pendant la grossesse, votre bébé peut avoir un poids inférieur à celui attendu à la naissance. Chez les femmes qui prennent du valproate, environ 11 à 15 bébés sur 100 peuvent avoir un poids inférieur à celui attendu à la naissance. En comparaison, cela concerne 5 à 10 bébés sur 100 chez les femmes de la population générale.
· Avant de vous prescrire ce médicament, votre médecin spécialiste devra vous avoir expliqué les risques pour votre bébé en cas de grossesse pendant la prise de valproate. Si vous envisagez une grossesse par la suite, vous ne devez pas arrêter de prendre votre médicament ou votre méthode de contraception avant d’en avoir parlé avec votre médecin.
· Si vous êtes un parent ou soignant d’une enfant de sexe féminin traitée par valproate, vous devez contacter le médecin spécialiste dès que l’enfant a ses premières règles.
· Certaines pilules contraceptives (pilules contenant des œstrogènes) peuvent abaisser les taux de valproate dans votre sang. Assurez-vous de discuter avec votre médecin de la méthode de contraception la plus appropriée pour vous.
Veuillez choisir la situation qui s’applique à votre cas parmi la liste ci-dessous et lire le paragraphe correspondant :
o JE COMMENCE UN TRAITEMENT PAR DEPAKINE
o JE PRENDS DEPAKINE ET JE NE PREVOIS PAS D’AVOIR UN ENFANT
o JE PRENDS DEPAKINE ET JE PREVOIS D’AVOIR UN ENFANT
o JE SUIS ENCEINTE ET JE PRENDS DEPAKINE
JE COMMENCE UN TRAITEMENT PAR DEPAKINE
S’il s’agit de votre première prescription de DEPAKINE, votre médecin spécialiste devra vous expliquer les risques du traitement pour l’enfant à naître en cas de grossesse. Si vous êtes en âge d’avoir des enfants, vous devez utiliser au moins une méthode de contraception efficace, sans interruption, pendant toute la durée de votre traitement par DEPAKINE. Pour obtenir des conseils sur la contraception, adressez-vous à votre médecin généraliste, votre gynécologue ou à un centre de planning familial.
Messages clés :
· Avant de débuter le traitement, votre médecin devra s’assurer qu’aucun autre traitement que le valproate n’est possible pour vous.
· Avant de débuter le traitement, votre médecin vous demandera d’effectuer un test de grossesse. Le résultat vu par votre médecin doit confirmer que vous n’êtes pas enceinte lorsque vous débutez le traitement par DEPAKINE.
· Vous devez utiliser au moins une méthode de contraception efficace (de préférence, un dispositif intra-utérin ou un implant contraceptif) ou deux méthodes efficaces qui fonctionnent différemment (par exemple, une pilule hormonale et un préservatif) pendant toute la durée de votre traitement par DEPAKINE.
· Vous devez discuter des méthodes de contraception appropriées avec votre médecin. Votre médecin vous donnera des informations sur la prévention d’une grossesse et pourra vous orienter vers un spécialiste qui vous donnera des conseils en matière de contraception.
· Vous devez consulter régulièrement (au moins une fois par an) un médecin spécialiste expérimenté dans le traitement de l’épilepsie. Lors de cette consultation, votre médecin s’assurera que vous êtes consciente des risques et que vous avez compris les informations liées aux risques du valproate pendant la grossesse.
· Si vous souhaitez avoir un enfant, parlez-en à votre médecin spécialiste avant d’arrêter votre contraception.
· Si vous êtes enceinte ou si vous pensez l'être, prenez rendez-vous en urgence avec votre médecin spécialiste expérimenté dans le traitement de l’épilepsie.
JE PRENDS DEPAKINE ET JE NE PREVOIS PAS D’AVOIR UN ENFANT
Si vous poursuivez le traitement par DEPAKINE et que vous ne prévoyez pas d’avoir un enfant, assurez-vous d’utiliser au moins une méthode de contraception efficace, sans interruption, pendant toute la durée du traitement par DEPAKINE. Pour obtenir des conseils sur la contraception, adressez-vous à votre médecin généraliste, votre gynécologue ou à un centre de planning familial.
Messages clés :
· Votre médecin spécialiste doit s’assurer régulièrement (au moins une fois par an) qu’aucun autre traitement que le valproate n’est possible pour vous.
· Vous devez utiliser au moins une méthode de contraception efficace (de préférence, un dispositif intra-utérin ou un implant contraceptif) ou deux méthodes efficaces qui fonctionnent différemment (par exemple, une pilule hormonale et un préservatif), pendant toute la durée de votre traitement par DEPAKINE.
· Vous devez discuter des méthodes de contraception appropriées avec votre médecin. Votre médecin vous donnera des informations sur la prévention d’une grossesse et pourra vous orienter vers un spécialiste qui vous donnera des conseils en matière de contraception.
· Vous devez consulter régulièrement (au moins une fois par an) un médecin spécialiste expérimenté dans le traitement de l’épilepsie. Lors de cette consultation, votre médecin s’assurera que vous êtes consciente des risques et que vous avez compris les informations liées aux risques du valproate pendant la grossesse.
· Si vous souhaitez avoir un enfant, parlez-en à votre médecin avant d’arrêter votre contraception.
· Si vous êtes enceinte ou si vous pensez l'être, prenez rendez-vous en urgence avec votre médecin spécialiste expérimenté dans le traitement de l’épilepsie.
JE PRENDS DEPAKINE ET JE PREVOIS D’AVOIR UN ENFANT
Les bébés nés de mères traitées par valproate présentent un risque grave de malformations et de troubles du développement qui peuvent se révéler lourdement handicapants. Si vous prévoyez d’avoir un enfant, prenez d’abord rendez-vous avec votre médecin spécialiste expérimenté dans le traitement de l’épilepsie.
N’arrêtez pas de prendre DEPAKINE ou votre contraception avant d'en avoir parlé avec votre médecin. Votre médecin vous fournira des conseils supplémentaires et vous orientera vers un médecin spécialiste expérimenté dans le traitement de l’épilepsie afin de pouvoir évaluer à temps les autres traitements possibles. Votre spécialiste pourra mettre en place différentes mesures pour que votre grossesse se déroule au mieux et que les risques pour vous et l'enfant à naître soient réduits autant que possible.
Votre spécialiste devra tout mettre en œuvre pour arrêter ce traitement par DEPAKINE, longtemps avant que vous ne deveniez enceinte, afin de s’assurer que votre maladie est stable. Pour les situations exceptionnelles où ce n’est pas possible, voir le paragraphe suivant (« JE SUIS ENCEINTE ET JE PRENDS DEPAKINE »).
Si vous envisagez une grossesse, interrogez votre médecin sur la prise d'acide folique. L’acide folique pourrait diminuer le risque général de spina bifida et de fausse couche précoce inhérent à toute grossesse. Néanmoins, il est peu probable qu’il diminue le risque de malformations liées à l’utilisation de valproate.
Messages clés :
· N’arrêtez pas de prendre DEPAKINE sans que votre médecin ne vous l’ait demandé.
· N'arrêtez pas d’utiliser vos méthodes de contraception avant d’en avoir discuté avec votre médecin spécialiste et convenu ensemble d’un traitement, ceci afin de s’assurer que votre maladie est sous contrôle et que les risques pour votre bébé sont réduits.
· Prenez d’abord rendez-vous avec votre médecin spécialiste. Lors de cette consultation, votre médecin s’assurera que vous êtes consciente des risques et que vous avez bien compris les informations liées aux risques du valproate pendant la grossesse.
· Votre médecin spécialiste devra tout essayer pour arrêter le traitement par DEPAKINE, longtemps avant que vous ne deveniez enceinte.
· Si vous êtes enceinte ou si vous pensez l'être, prenez rendez-vous en urgence avec votre médecin spécialiste expérimenté dans le traitement de l’épilepsie.
JE SUIS ENCEINTE ET JE PRENDS DEPAKINE
Les bébés nés de mères traitées par le valproate présentent un risque grave de malformations et de troubles du développement intellectuel et moteur et du comportement qui peuvent se révéler lourdement handicapants. N'arrêtez pas de prendre DEPAKINE sans que votre médecin vous l’ait demandé cela pourrait aggraver votre maladie. Si vous êtes enceinte ou pensez l’être, prenez rendez-vous en urgence avec votre médecin spécialiste expérimenté dans le traitement de l’épilepsie :
· il vous donnera des conseils supplémentaires,
· il devra tout essayer pour arrêter le traitement, et évaluer l’ensemble des autres thérapeutiques possibles.
Dans des situations exceptionnelles, si DEPAKINE est l’unique option thérapeutique disponible pendant votre grossesse :
· Votre médecin pourra vous orienter vers un spécialiste afin que vous et votre partenaire receviez de l’aide et des conseils concernant une grossesse sous valproate.
· Votre médecin spécialiste essaiera de diminuer la dose prescrite.
· Vous serez suivie étroitement, à la fois pour le traitement de votre maladie et la surveillance du développement de l’enfant à naître.
· Interrogez votre médecin sur la prise d’acide folique. L'acide folique pourrait diminuer le risque général de spina bifida et de fausse couche précoce possible au cours de toute grossesse. Néanmoins, les données disponibles ne montrent pas qu'il diminue le risque de malformations liées à l’utilisation de valproate.
· Avant l’accouchement : votre médecin vous prescrira certaines vitamines pour éviter que ce médicament ne provoque des saignements durant les premiers jours de vie ou des troubles dans la formation des os de votre bébé.
· Après l’accouchement : une injection de vitamine K pourra également être prescrite à votre bébé, à la naissance, pour éviter des saignements.
· Chez l’enfant : prévenez le(s) médecin(s) qui suivra(ont) votre enfant que vous avez été traitée par valproate pendant votre grossesse. Il(s) mettra(ont) en place un suivi rapproché du développement neurologique de votre enfant afin de lui apporter des soins spécialisés le plus tôt possible, si nécessaire.
Messages clés :
· Si vous êtes enceinte ou si vous pensez l’être, prenez rendez-vous en urgence avec votre médecin spécialiste expérimenté dans le traitement de l’épilepsie.
· N'arrêtez pas de prendre DEPAKINE sans que votre médecin spécialiste ne vous l’ait demandé.
· Votre médecin spécialiste expérimenté dans le traitement de l’épilepsie doit évaluer toutes les possibilités pour arrêter ce traitement.
· Votre médecin spécialiste doit vous donner des informations complètes sur les risques liés à la prise de DEPAKINE pendant la grossesse, notamment les risques de malformations (malformations congénitales) et les troubles du développement (intellectuel, moteur, comportemental) des enfants.
· Assurez-vous d’être orientée vers un médecin spécialisé en surveillance prénatale afin de détecter d’éventuelles malformations.
· Prévenez les médecins qui suivront votre enfant que vous avez pris DEPAKINE pendant votre grossesse, ils mettront en place un suivi rapproché de son développement neurologique.
Informations importantes à l'attention des adolescents et hommes en âge d’avoir des enfants
Risques potentiels liés à la prise de valproate dans les 3 mois précédant la conception d’un enfant
Une étude suggère un risque potentiel de troubles du développement mental et/ou moteur (problèmes de développement dans la petite enfance) chez les enfants dont le père a été traité par valproate dans les 3 mois précédant la conception. Dans cette étude, environ 5 % des enfants nés de pères traités par valproate ont présenté des troubles du développement, alors que dans le groupe de comparaison dans lequel les pères étaient traités par d’autres médicaments, lamotrigine ou lévétiracétam, environ 3 % des enfants présentaient de tels troubles. Le risque n’est pas connu pour les enfants nés de pères ayant arrêté le traitement par valproate plus de 3 mois avant la conception (temps nécessaire pour former de nouveaux spermatozoïdes). L’étude présente des limites, il n’est donc pas certain que l’augmentation du risque de troubles du développement moteur et mental suggéré par cette étude soit causée par le valproate. Le nombre de patients inclus dans l’étude n’était pas suffisant pour déterminer les types particuliers de troubles du développement moteur et mental que les enfants sont susceptibles de développer.
Par mesure de précaution, votre médecin discutera avec vous :
· Du risque potentiel de troubles du développement chez les enfants nés de pères traités par valproate.
· De la nécessité d’envisager une contraception efficace pour vous et votre partenaire pendant le traitement et pendant les 3 mois après l’arrêt du valproate.
· De la nécessité de consulter votre médecin lorsque vous prévoyez de concevoir un enfant et avant d’arrêter la contraception.
· De la possibilité d’envisager d’autres traitements plus appropriés pour traiter votre maladie, en fonction de votre situation individuelle.
Vous ne devez pas faire de don de sperme au cours de votre traitement avec le valproate ou l’un de ses dérivés, et au moins trois mois après l’arrêt de celui-ci.
Si vous envisagez d’avoir un enfant, parlez-en à votre médecin.
Si votre partenaire débute une grossesse alors que vous preniez du valproate dans les 3 mois précédant la conception et si vous avez des questions, contactez votre médecin. N’arrêtez pas votre traitement sans en parler à votre médecin car si vous l’arrêtez seul, cela vous exposerait à la réapparition de vos symptômes.
Vous devez consulter régulièrement votre médecin. Pendant cette consultation, votre médecin discutera avec vous des précautions associées à l’utilisation du valproate et de la possibilité d’autres traitements pour votre maladie, selon votre situation individuelle.
Lisez la brochure d’information patient qui vous sera remise par votre médecin. Une carte patient vous sera également remise par votre pharmacien pour vous rappeler les risques potentiels du valproate.
Allaitement
Vous ne devez pas allaiter si vous prenez ce médicament sauf avis contraire de votre médecin.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
DEPAKINE peut provoquer une somnolence, en particulier si vous prenez en même temps un autre médicament anticonvulsivant ou pouvant augmenter la somnolence.
Si vous ressentez cet effet et si votre maladie n’est pas encore contrôlée et que vous continuez à avoir des crises, vous ne devez pas conduire un véhicule ou utiliser une machine.
DEPAKINE 200 mg/ml, solution buvable contient du sodium
Ce médicament contient 28 mg de sodium (composant principal du sel de cuisine/table) pour 200 mg de valproate de sodium. Cela équivaut à 1,4 % de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte. Vous devez en tenir compte si vous suivez un régime sans sel ou pauvre en sel.
3. COMMENT PRENDRE DEPAKINE 200 mg/ml, solution buvable ?
Instructions pour un bon usage
Filles et femmes en âge d’avoir des enfants
Le traitement par DEPAKINE doit être instauré et surveillé par un médecin spécialiste dans le traitement de l’épilepsie. Le traitement ne doit pas être prescrit chez les filles, adolescentes, femmes en âge d’avoir des enfants sauf en cas d’inefficacité ou d’intolérance aux autres traitements. Si aucun autre traitement n’est possible, le valproate vous sera prescrit et dispensé sous des conditions très strictes (indiquées dans le programme de prévention de la grossesse). Un spécialiste doit réévaluer au moins une fois par an la nécessité du traitement.
Adolescents et hommes en âge d’avoir des enfants
Il est recommandé que le traitement par DEPAKINE soit instauré et surveillé par un médecin spécialiste dans la prise en charge de l’épilepsie - voir rubrique 2, paragraphe « Informations importantes à l'attention des adolescents et hommes en âge d’avoir des enfants ».
Posologie
· La dose quotidienne à utiliser est déterminée et contrôlée individuellement par votre médecin.
· Votre médecin doit vous prescrire une dose en milligrammes (mg) et non en millilitres (ml). Cette information est importante car la seringue utilisée pour prélever la dose appropriée dans le flacon est graduée en milligrammes (mg). Si votre prescription a été rédigée en millilitres (ml), contactez votre médecin ou votre pharmacien.
· La dose est habituellement répartie en
o 2 prises par jour chez les enfants âgés de moins de 1 an.
o 3 prises par jour, chez les adultes et les enfants âgés de plus de 1 an.
· Prenez votre dose de préférence au cours de vos repas.
Patients atteints de troubles rénaux
Votre médecin peut décider d’ajuster votre dose.
Mode d’administration
|
|
La solution buvable sera absorbée après dilution dans un peu de boisson non gazeuse. Le flacon de solution buvable est accompagné d’une seringue pour administration orale (piston rose) uniquement réservée à l’utilisation de ce médicament. Cette seringue pour administration orale est graduée de 50 mg à 400 mg, tous les 50 mg avec des graduations secondaires de 25 mg, et elle porte la mention « DEPAKINE 200 mg/ml, solution buvable ». Administrez la solution buvable uniquement avec la seringue pour administration orale présente dans cette boîte. Les traits de graduation indiquent les doses exprimées en milligrammes (mg).
|
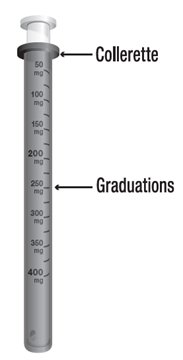

|
Ouverture du flacon Pour ouvrir le flacon, tourner le bouchon sécurité-enfant en appuyant sur le dessus.
|
|
Utilisation de la seringue pour administration orale
· La dose à administrer est obtenue en tirant le piston jusqu’à la graduation correspondant à la quantité en milligrammes (mg) prescrite par votre médecin.
· La dose est lue au niveau de la collerette de la seringue pour administration orale.
Exemple pour une dose de 200 mg
|
1) Remplir la seringue pour administration orale en tirant le piston.
2) Ajuster la dose à 200 mg.
3) Vider le contenu de la seringue pour administration orale dans un verre et le diluer dans un peu de boisson non gazeuse.
|
|
|
|
Après utilisation, refermer le flacon, bien rincer à l’eau et sécher la seringue pour administration orale. Puis ranger immédiatement la seringue pour administration orale au sein de sa boîte dans un endroit inaccessible aux enfants. Ne jamais séparer la seringue pour administration orale des autres éléments de conditionnement du médicament (boîte, notice). |
|
|
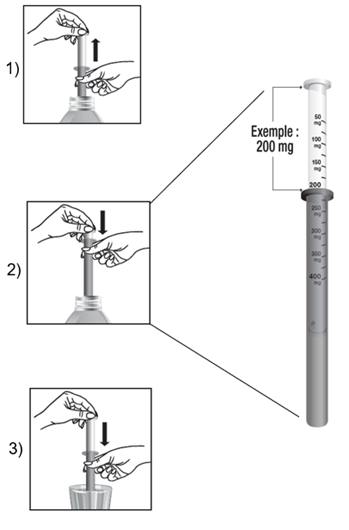
Durée du traitement
N’interrompez pas la prise de ce médicament sans avis médical.
Si vous avez pris plus de DEPAKINE 200 mg/ml, solution buvable que vous n’auriez dû
Consultez immédiatement votre médecin ou les urgences médicales.
Si vous oubliez de prendre DEPAKINE 200 mg/ml, solution buvable
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre DEPAKINE 200 mg/ml, solution buvable
N’arrêtez pas la prise de DEPAKINE sans l’avis de votre médecin. L’interruption de votre traitement devra être réalisée de manière progressive. Si vous arrêtez de prendre DEPAKINE brutalement ou avant que votre médecin ne vous l’ait demandé, vous vous exposez à un risque accru de crises.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Informez immédiatement votre médecin si vous remarquez l’un des effets indésirables graves suivants ; vous pourriez avoir besoin de soins médicaux urgents :
· des problèmes d’équilibre et de coordination, une sensation de léthargie ou de moins bonne vigilance, associés à des vomissements. Cela peut être dû à une augmentation de la quantité d'ammonium dans votre sang,
· une atteinte du foie (hépatite) ou du pancréas (pancréatite), pouvant être grave et mettre votre vie en danger et qui peut commencer soudainement par une fatigue, une perte d’appétit, un abattement, une somnolence, des nausées, des vomissements, des douleurs dans le ventre.
· une réaction allergique
o brusque gonflement du visage et/ou du cou pouvant entraîner une difficulté à respirer et vous mettre en danger (œdème de Quincke),
o réaction allergique grave (syndrome d’hypersensibilité médicamenteuse) associant plusieurs symptômes tels que de la fièvre, une éruption sur la peau, une augmentation de la taille des ganglions, une atteinte du foie, du rein, et des anomalies des examens sanguins telles qu'une augmentation du nombre de certains globules blancs (éosinophiles).
· une éruption de boutons sur la peau avec parfois des bulles pouvant aussi affecter la bouche (érythème polymorphe), une éruption de bulles avec décollement de la peau pouvant s’étendre rapidement à tout le corps et vous mettre en danger (syndrome de Lyell, syndrome de Stevens-Johnson),
· des difficultés à respirer, une douleur ou pression dans la poitrine (en particulier lors de l’inspiration), essoufflement et toux sèche en raison de l’accumulation de liquide autour des poumons (épanchement pleural).
Autres effets indésirables possibles
· malformations congénitales et troubles du développement intellectuel et moteur (voir rubrique 2 - paragraphe « Grossesse, allaitement et fertilité »).
Informez immédiatement votre médecin ou votre pharmacien si l’un des effets indésirables suivants devient grave ou dure plus de quelques jours : vous pourriez avoir besoin d’un traitement médical urgent :
Très fréquent (touchant plus de 1 personne sur 10) :
· nausées,
· tremblements.
Fréquent (touchant jusqu’à 1 personne sur 10) :
· en début de traitement vomissements, douleurs à l’estomac, diarrhées,
· prise de poids,
· maux de tête,
· somnolence,
· convulsions,
· troubles de la mémoire,
· confusion, agressivité, agitation, troubles de l’attention, hallucinations (voir, entendre ou sentir des choses qui n’existent pas),
· troubles extrapyramidaux (ensemble de symptômes tels que des tremblements, une rigidité des membres et des difficultés pour marcher)*,
· fuite urinaire (incontinence urinaire),
· mouvements des yeux rapides et incontrôlables,
· perte de l'audition,
· affections de la gencive (troubles gingivaux), en particulier augmentation du volume de la gencive (hypertrophie gingivale),
· bouche douloureuse, enflée, aphtes et sensation de brûlure de la bouche (stomatite),
· chute des cheveux,
· troubles des règles (irrégularité menstruelle),
· saignements,
· sensations nauséeuses ou vertigineuses,
· troubles de l’ongle et du lit de l’ongle,
· diminution du nombre des plaquettes (thrombopénie), diminution du nombre de globules rouges (anémie),
· diminution de la quantité de sodium dans le sang (hyponatrémie, syndrome de sécrétion inappropriée de l’hormone antidiurétique).
Peu fréquent (touchant jusqu’à 1 personne sur 100) :
· troubles de la vigilance, pouvant aller jusqu'au coma passager, qui régresse après diminution de la dose ou arrêt du traitement,
· difficultés à coordonner ses mouvements,
· syndrome parkinsonien réversible*,
· engourdissement ou fourmillement des mains et des pieds,
· texture anormale des cheveux, changements de la couleur des cheveux, pousse anormale des cheveux,
· éruption de boutons ou de plaques sur la peau,
· pilosité excessive, particulièrement chez les femmes, virilisme, acné (hyperandrogénie),
· baisse de la température corporelle (hypothermie),
· gonflements des extrémités (œdèmes),
· aménorrhée (absence de règles),
· augmentation du nombre et de la gravité des convulsions, apparition de crises convulsives de type différent,
· difficulté respiratoire et douleur dues à l’inflammation de la membrane protectrice des poumons (épanchement pleural),
· diminution de l’ensemble des cellules du sang : globules blancs, globules rouges et plaquettes (pancytopénie),
· diminution sévère du nombre de globules blancs (leucopénie) observée lors d’analyses de sang, parfois révélée par de la fièvre et une difficulté à respirer,
· des cas de troubles osseux se manifestant par une fragilisation des os (ostéopénie), une diminution de la masse osseuse (ostéoporose) et des fractures ont été rapportés. Consultez votre médecin ou votre pharmacien en cas de traitement de longue durée par un médicament antiépileptique, d’antécédent d’ostéoporose ou de prise de corticostéroïdes,
· inflammation des vaisseaux sanguins.
Rare (touchant jusqu’à 1 personne sur 1 000) :
· troubles de la fertilité masculine généralement réversibles après au moins 3 mois d’arrêt du traitement et possiblement réversibles après diminution de la dose. N’arrêtez pas votre traitement sans en avoir d’abord parlé à votre médecin,
· fonctionnement anormal des ovaires (syndrome des ovaires polykystiques),
· troubles du comportement, augmentation de l’activité psychomotrice, difficultés d’apprentissage,
· réaction auto-immune avec douleur des articulations, éruptions sur la peau et fièvre (lupus érythémateux disséminé),
· diminution de l’activité de la glande thyroïde (hypothyroïdie),
· douleurs musculaires, faiblesse musculaire pouvant être graves (rhabdomyolyse),
· obésité,
· émission involontaire d’urine le plus souvent la nuit (énurésie),
· atteinte des reins (insuffisance rénale, néphrite tubulo-interstitielle) pouvant se manifester par une diminution du débit urinaire,
· uriner beaucoup et avoir soif (syndrome de Fanconi),
· augmentation du volume des globules rouges (macrocytose), diminution importante du nombre de globules blancs (agranulocytose),
· appauvrissement de la production des cellules sanguines (aplasie médullaire), anomalie de la production des cellules sanguines (myélodysplasie), observées lors d’analyses de sang, parfois révélées par de la fièvre et une difficulté à respirer,
· diminution des facteurs de coagulation, anomalies des tests de coagulation (augmentation de l’INR, allongement du TCA...),
· diminution de la quantité de vitamine B8 (biotine)/de biotinidase,
· augmentation de la quantité d’ammonium dans le sang,
· vision double,
· troubles de la mémoire et des capacités mentales d’apparition progressive (troubles cognitifs, syndrome démentiel)*. Ces troubles diminuent quelques semaines à quelques mois après l’arrêt du traitement.
Fréquence indéterminée (la fréquence ne peut pas être estimée à partir des données disponibles) :
· diminution du taux de carnitine (observée dans les analyses sanguines ou musculaires),
· zones plus sombres sur la peau et les muqueuses (hyperpigmentation).
N'arrêtez pas votre traitement sans en avoir d’abord parlé à votre médecin,
* Ces symptômes peuvent être associés à des signes radiologiques au niveau du cerveau (atrophie cérébrale).
Effets secondaires supplémentaires chez les enfants
Certains effets secondaires du valproate surviennent plus fréquemment chez les enfants ou sont plus graves que chez les adultes. Ceux-ci entrainent une atteinte du foie, une inflammation du pancréas (pancréatite), une agressivité, une agitation, une perturbation de l’attention, un comportement anormal, une hyperactivité et un trouble d’apprentissage.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet https: //signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER DEPAKINE 200 mg/ml, solution buvable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage. La date de péremption fait référence au dernier jour de ce mois.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient DEPAKINE 200 mg/ml, solution buvable
· La substance active est :
Valproate de sodium............................................................................................................ 20,00 g
Pour 100 ml.
1 ml de solution correspond à 200 mg de valproate de sodium.
· Les autres composants sont :
Urée, solution d'hydroxyde de sodium à 30 % et eau purifiée.
Qu’est-ce que DEPAKINE 200 mg/ml, solution buvable et contenu de l’emballage extérieur
Ce médicament se présente sous forme de solution buvable. Flacon de 40 ml.
Titulaire de l’autorisation de mise sur le marché
82, AVENUE RASPAIL
94250 GENTILLY
Exploitant de l’autorisation de mise sur le marché
SANOFI WINTHROP INDUSTRIE
82, AVENUE RASPAIL
94250 GENTILLY
1-3, ALLEE DE LA NESTE
31770 COLOMIERS
ou
SANOFI WINTHROP INDUSTRIE
30-36 AVENUE GUSTAVE EIFFEL
37100 TOURS
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
[QR Code]
Des informations détaillées et actualisées sont disponibles en scannant ce code QR ou à l'adresse : www.ansm.sante.fr/Traitement-par-Valproate
CONSEILS GENERAUX
L’épilepsie est une maladie neurologique. Elle est l’expression d’un fonctionnement anormal, aigu et transitoire de l’activité électrique du cerveau, se traduisant par des crises épileptiques. Les crises peuvent se répéter pendant un certain temps de la vie d’un individu.
Les formes d’expression des crises et leur évolution sont multiples : il n’y a pas une mais des épilepsies.
De même, il n’y a pas un traitement mais des traitements : votre médecin vous prescrira celui qui est le mieux adapté à votre cas.
Pour que le médicament qui vous a été prescrit soit efficace, vous devez impérativement suivre les recommandations de votre médecin et respecter :
· la dose journalière prescrite,
· l’horaire des prises,
· la durée du traitement, généralement prolongée,
· les conseils d’hygiène de vie : évitez le surmenage, le manque de sommeil ainsi que l’alcool.
La modification des doses et, surtout, l’arrêt brutal du traitement peuvent entraîner la réapparition des troubles.
N'OUBLIEZ PAS VOTRE MEDICAMENT SI VOUS PARTEZ EN VOYAGE.