Base de données publique
des médicaments
Visiter [medicaments.gouv.fr] ![Visiter [medicaments.gouv.fr]](/img/icone_lien.png "Visiter [medicaments.gouv.fr] - nouvelle fenêtre")



DYSPORT 500 UNITES SPEYWOOD, poudre pour solution injectable - Résumé des caractéristiques du produit |
 |
|
ANSM - Mis à jour le : 22/06/2022
DYSPORT 500 UNITES SPEYWOOD, poudre pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE 
Toxine botulinique de type A (complexe toxine-hémagglutinine BoTX-A).................. 500 U Speywood
Pour un flacon
Les unités Speywood de DYSPORT sont spécifiques à la préparation et ne sont pas interchangeables avec d’autres préparations de toxine botulinique.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour solution injectable.
4.1. Indications thérapeutiques
· Blépharospasme.
· Spasme hémifacial.
· Torticolis spasmodique.
· Traitement symptomatique local de la spasticité (hyperactivité musculaire) des membres supérieurs et/ou inférieurs.
· Traitement de l'incontinence urinaire chez les adultes avec une hyperactivité neurologique du détrusor due à une blessure médullaire (traumatique ou non traumatique) ou à une sclérose en plaques, qui effectuent régulièrement un sondage intermittent propre.
Enfants à partir de 2 ans
· Traitement symptomatique local de la spasticité des membres supérieurs et/ou inférieurs.
Ce traitement médicamenteux doit être inclus dans une prise en charge globale multidisciplinaire (associant neurologue, pédiatre, médecin de médecine physique et de réadaptation, chirurgien orthopédiste…).
NB : DYSPORT 500 UNITES SPEYWOOD DOIT ETRE ADMINISTRE PAR DES MEDECINS AYANT DEJA UNE BONNE EXPERIENCE DE L’UTILISATION DE LA TOXINE DANS CES INDICATIONS.
4.2. Posologie et mode d'administration
Les doses recommandées de DYSPORT 500 UNITES SPEYWOOD sont spécifiques de la préparation et ne sont pas interchangeables avec les autres préparations de toxines botuliniques A. Elles sont exprimées en unités SPEYWOOD (voir rubrique 4.4).
Un intervalle minimum de 12 semaines entre deux séances d’injection doit être respecté.
DYSPORT doit être uniquement administré par des médecins ayant été formés de façon appropriée.
Une aiguille stérile de 23 ou 25 gauge doit être utilisée pour réaliser l’injection.
En cas d’antécédents d’atteinte neurogène de la face, il est recommandé de réduire la posologie (voir rubrique 4.4).
Les instructions pour la reconstitution sont spécifiques à chaque dosage de DYSPORT (DYSPORT 300 Unités Speywood et DYSPORT 500 Unités Speywood). Les volumes de dilution correspondent à des concentrations qui sont spécifiques à chaque indication.
|
Concentration en unités Speywood par ml |
Volume de solution injectable NaCl 0,9%* à ajouter par flacon de DYSPORT 500 Unités |
|
500 Unités/ml |
1 ml |
|
200 Unités/ml |
2,5 ml |
|
100 Unités/ml |
5 ml |
*stérile, sans conservateur
Pour la spasticité des membres inférieurs chez l’enfant, la dose étant déterminée en fonction du poids corporel, une dilution supplémentaire peut être nécessaire afin d’atteindre le volume final pour l’injection.
BLEPHAROSPASME ET SPASME HEMIFACIAL
Posologie
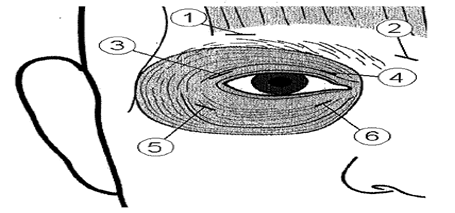
Dans le traitement du blépharospasme, la toxine botulinique est habituellement injectée dans les muscles suivants : orbicularis oculi, corrugator et procerus. Le traitement standard implique 4 sites d’injection autour de chacun des yeux dont deux injections au niveau des paupières supérieures, l’une médiane et l’autre latérale à proximité du canthus.
Dans le traitement du spasme hémifacial, les muscles injectés par la toxine botulinique sont très similaires à ceux injectés pour traiter un blépharospasme et incluent le plus souvent l’orbicularis oculi et le corrugator (parmi d’autres muscles de la face).
Habituellement, lors de la première séance d’injection pour le traitement d’un spasme hémifacial, seules les régions peri-oculaires sont injectées.
Même si le spasme hémifacial affecte la région inférieure de la face, seuls les muscles de la région supérieure de la face sont injectés lors de la première séance car il est admis que cela peut être suffisant pour contrôler les spasmes affectant la région inférieure de la face.
Une étude clinique de recherche de doses pour le traitement du blépharospasme essentiel et bénin a montré que l’injection d’une dose de 40 unités de DYSPORT par œil est significativement efficace. L’injection d’une dose de 80 unités par œil engendre un effet de plus longue durée. Cependant, l’incidence des évènements indésirables et plus particulièrement du ptosis est dose-dépendant. Lors du traitement d’un blépharospasme ou d’un spasme hémifacial, la dose maximale injectée ne doit pas excéder une dose totale de 120 unités par œil.
Une injection de 10 unités (0,05 ml) dans la partie interne (médiane) et de 10 unités (0,05 ml) dans la partie externe (latérale) de la jonction entre les zones préseptales et orbitales de chacun des deux muscles orbiculaires, supérieur (3 et 4) et inférieur (5 et 6), de chaque œil doit être réalisée. Afin de réduire le risque de ptosis, les injections à proximité du muscle élévateur de la paupière supérieure doivent être évitées.
|
|
Lors de l’injection dans la paupière supérieure, l’aiguille doit être orientée de telle sorte que l’on évite le centre de la paupière, partie où s’insère le muscle releveur de la paupière. Le diagramme ci-dessus aide à repérer les points d’injection.
Les injections doivent être renouvelées approximativement toutes les 12 semaines ou dès que nécessaire pour éviter la réapparition des symptômes, mais elles ne doivent pas être répétées plus fréquemment que toutes les 12 semaines.
Pour les injections ultérieures, si la réponse observée à la dose initiale est insuffisante, la dose injectée par œil peut être augmentée comme suit : 60 unités (10 unités (0,05 ml) dans la partie interne et 20 unités (0,1 ml) dans la partie externe) ; 80 unités (20 unités (0,1 ml) dans la partie interne et 20 unités (0,1 ml) dans la partie externe ou jusqu’à 120 unités (20 unités (0,1 ml) dans la partie interne et 40 unités (0,2 ml) dans la partie externe) au-dessus et en-dessous de chaque œil selon la manière décrite précédemment. D’autres sites, localisés dans le muscle frontalis, au-dessus du sourcil (1 et 2) peuvent également être injectés si les spasmes gênent la vision.
Dans le cas de blépharospasme unilatéral, les injections devront être limitées à l’œil atteint. Les patients atteints de spasme hémifacial doivent être traités de la même façon que lors de la prise en charge d’un blépharospasme unilatéral. Les doses recommandées sont applicables à l’adulte quel que soit son âge y compris le sujet âgé.
Population pédiatrique
Chez l’enfant, la tolérance et l’efficacité de DYSPORT dans le traitement du blépharospasme et du spasme hémifacial chez l’enfant n’ont pas été démontrées.
Mode d’administration
A l'aide d'une aiguille stérile, introduire dans le flacon 2,5 ml d’une solution injectable de chlorure de sodium à 0,9 pour cent (voir rubrique 6.6). On obtient ainsi une solution limpide contenant 200 UNITES SPEYWOOD/ml de substance active.
DYSPORT est administré par injection par voie sous-cutanée dans les parties interne (médiane) et externe (latérale) de la jonction entre les zones préseptales et orbitales de chacun des deux muscles orbiculaires (supérieur et inférieur) des yeux.
TORTICOLIS SPASMODIQUE
Posologie
Les doses recommandées pour le traitement du torticolis sont applicables à l’adulte, quel que soit son âge, à condition qu’il présente un poids normal et qu’il ne présente pas de signe de réduction de la masse musculaire du cou. L’injection d’une dose plus faible est recommandée chez le patient présentant un poids sensiblement insuffisant ou chez le sujet âgé, chez lequel peut exister une réduction de la masse musculaire.
La dose initiale recommandée est de 500 unités par patient (soit 1 ml, pour la dilution de 1 flacon de 500 unités dans 1 ml).
Cette dose ne doit jamais excéder 1000 unités par patient et par séance d’injection.
La dose totale doit être répartie entre les 2 ou 3 muscles cervicaux les plus actifs (le plus souvent : sterno-cléido-mastoïdien, splenius, trapèze ou angulaire).
Afin de minimiser le risque de dysphagie, le muscle sterno-cléido-mastoïdien ne doit pas être injecté de façon bilatérale et la dose initiale pour ce muscle ne doit pas dépasser 150 unités (soit 0,3 ml).
Le clinicien est libre de déterminer avec ou sans guidage électromyographique (E.M.G), les muscles les plus actifs, et le nombre de sites à injecter par muscle.
Pour chaque muscle, la dose sera répartie en 2 ou 3 sites.
Lors des injections ultérieures, la dose doit être adaptée en fonction de la réponse clinique et des effets indésirables observés. L’injection de doses comprises entre 250 et 1000 unités est recommandée bien que l’injection des doses les plus fortes puisse être associée à une augmentation de la survenue d’effets indésirables, en particulier la dysphagie.
Les séances d’injection doivent être répétées environ toutes les 16 semaines ou dès que nécessaire pour maintenir l’effet recherché, mais pas plus fréquemment que toutes les 12 semaines.
En cas de forme rotationnelle de torticolis : répartir la dose de 500 unités en injectant 350 unités dans le muscle splénius de la tête, de façon ipsilatérale à la rotation, dans la direction de l’axe menton/tête, et 150 unités dans le muscle sternomastoïdien, de façon controlatérale à l’axe de rotation.
En cas de laterocolis, répartir la dose de 500 unités en injectant 350 unités dans le splénius ipsilatéral de la tête et 150 unités dans le sternomastoïdien ipsilatéral. Si une élévation de l’épaule est associée, une injection dans le trapèze ipsilatéral ou dans le muscle angulaire de l’omoplate pourrait être requise dans les cas d’hypertrophie visible du muscle ou des résultats de l’électromyogramme.
Quand l’injection de 3 muscles est nécessaire, répartir la dose de 500 unités comme suit : 300 unités dans le muscle splénius de la tête, 100 unités dans le sternomastoïdien et 100 unités dans le troisième muscle.
En cas de retrocolis, répartir la dose de 500 unités en injectant 250 unités dans chacun des splénius de la tête. Les injections bilatérales dans le muscle splénius de la tête peuvent augmenter le risque de faiblesse musculaire au niveau du cou.
Dans les autres formes de torticolis, l’identification des muscles les plus actifs est très dépendante de l’expertise du spécialiste et de l’EMG. L’EMG devrait être utilisé dans le diagnostic de toutes les formes complexes de torticolis, lors d’une réévaluation après échec des injections dans les formes non complexes de torticolis et pour guider les injections dans les muscles profonds ou chez les patients en surpoids chez lesquels les muscles du cou sont peu palpables.
Population pédiatrique
Chez l’enfant, la tolérance et l’efficacité de DYSPORT dans le traitement du torticolis chez l’enfant n’ont pas été démontrées.
Mode d’administration
A l’aide d’une aiguille stérile, introduire dans le flacon 1 ml d’une solution injectable de chlorure de sodium à 0,9 pour cent (voir rubrique 6.6). On obtient ainsi une solution limpide contenant 500 UNITES SPEYWOOD/ml de substance active.
DYSPORT est injecté par voie intramusculaire.
TRAITEMENT SYMPTOMATIQUE LOCAL DE LA SPASTICITE DES MEMBRES SUPERIEURS ET/OU INFERIEURS CHEZ L’ADULTE
Posologie
Spasticité du membre inférieur chez l’adulte
Des doses allant jusqu’à 1500 unités peuvent être administrées au cours d’une seule séance de traitement. La dose à l’initiation ou lors d’une nouvelle séance d’injection doit être adaptée à chaque patient en fonction de la taille, du nombre de muscles concernés, de la sévérité de la spasticité, de l’existence d’une faiblesse musculaire localisée et de la réponse du patient au traitement précédent. Cependant, la dose totale ne doit pas excéder 1500 unités.
Le volume administré à chaque site d’injection ne devrait généralement pas excéder 1ml.
|
Muscle |
Dose1 de DYSPORT recommandée (Unité Speywood) |
Nombre de sites2 d’injection recommandé |
|
Distal |
|
|
|
Soleus (soléaire) |
300-550 |
2 - 4 |
|
Gastrocnemius (gastrocnémien) |
|
|
|
Chef médial |
100-450 |
1 - 3 |
|
Chef latéral |
100-450 |
1 - 3 |
|
Tibialis posterior (tibial postérieur) |
100-250 |
1 - 3 |
|
Flexor digitorum longus (fléchisseur commun des orteils) |
50-200 |
1 - 2 |
|
Flexor digitorum brevis (court fléchisseur des orteils) |
50-200 |
1 - 2 |
|
Flexor hallucis longus (long fléchisseur de l’hallux) |
50-200 |
1 - 2 |
|
Flexor hallucis brevis (court fléchisseur de l’hallux) |
50-100 |
1 - 2 |
|
Proximal |
|
|
|
Rectus femoris (muscle droit fémoral) |
100-400 |
1 - 3 |
|
Muscle ischio-jambier |
100-400 |
1 - 3 |
|
Adductor magnus (grand adducteur) |
100-300 |
1 – 3 |
|
Adductor Longus (long adducteur) |
50-150 |
1 - 2 |
|
Adductor Brevis (court adducteur) |
50-150 |
1 - 2 |
|
Gracilis |
100-200 |
1 - 3 |
|
Gluteus maximus (grand fessier) |
100-400 |
1 - 2 |
|
1 Il conviendra d’utiliser une dose initiale moindre pour éviter l’apparition d’une faiblesse musculaire excessive des muscles concernés, par exemple chez les patients dont les muscles à traiter sont peu développés ou encore chez les patients qui nécessitent une injection concomitante dans un autre groupe musculaire. 2 le nombre de sites dépend du volume du muscle injecté |
||
Spasticité du membre supérieur chez l’adulte
La dose maximale recommandée ne doit pas dépasser 1000 unités (ou 1500 unités en cas d’injection dans les muscles de l’épaule).
La dose à l’initiation ou lors d’une nouvelle séance d’injection doit être adaptée à chaque patient en fonction de la taille, du nombre et de la localisation des muscles concernés, de la sévérité de la spasticité, de l’existence d’une faiblesse musculaire localisée, de la réponse du patient au traitement précédent et/ou d’antécédents d’évènements indésirables. Dans les essais cliniques, lors d’une séance d’injection, les doses de 500, 1000 et 1500 Unités Speywood ont été réparties entre les différents muscles sélectionnés (cf. tableau ci-après). A partir du deuxième cycle de traitement, des doses supérieures à 1000 Unités et jusqu’à 1500 Unités peuvent être administrées en cas d’injection dans les muscles de l’épaule. Dans le cas de l’administration d’une dose de 1500 U dans le membre supérieur, 500 U seront injectées dans les muscles de l’épaule.
Le volume administré à chaque site d’injection ne devrait généralement pas excéder 1ml. Des doses supérieures à 1500 Unités de DYSPORT n’ont pas été évaluées pour le traitement de la spasticité des membres supérieurs chez l’adulte.
|
Muscle |
Dose1 de DYSPORT recommandée (Unité Speywood) |
Nombre de sites2 d’injection recommandé |
|
Flexor carpi radialis (fléchisseur radial du carpe) |
25-200 U |
1 - 2 |
|
Flexor carpi ulnaris (fléchisseur ulnaire du carpe) |
25-200 U |
1 - 2 |
|
Flexor digitorum profundus (fléchisseur profond des doigts) |
100-200 U |
1 - 2 |
|
Flexor digitorum superficialis (fléchisseur superficiel des doigts) |
100-200 U |
1 - 2 |
|
Flexor Pollicis Longus (long fléchisseur du pouce) |
20-200 U |
1 |
|
Adductor Pollicis (adducteur du pouce) |
25-50 U |
1 |
|
Brachialis (brachial) |
50-400 U |
1 - 2 |
|
Brachioradialis (brachio-radial) |
50-200 U |
1 - 2 |
|
Biceps brachii (biceps brachial) |
50-400 U |
1 - 2 |
|
Pronator Teres (rond pronateur) |
45-200 U |
1 |
|
Triceps brachii, caput longum (Triceps brachial, chef long) |
150-300 U |
2 |
|
Pectoralis Major (Grand pectoral) |
100-300 U |
2 |
|
Subscapularis (Sous-scapulaire) |
75-300 U |
1 - 2 |
|
Latissimus Dorsi (Grand dorsal) |
150-300 U |
1 - 2 |
|
1 Il conviendra d’utiliser une dose initiale moindre pour éviter l’apparition d’une faiblesse musculaire excessive des muscles concernés, par exemple chez les patients dont les muscles à traiter sont peu développés ou encore chez les patients qui nécessitent une injection concomitante dans un autre groupe musculaire. 2 le nombre de sites dépend du volume du muscle injecté |
||
Le renouvellement de l’injection doit être réalisé lorsque l’effet clinique lié à la précédente injection diminue mais en respectant un intervalle minimum de 12 semaines après la précédente injection.
Dans les études cliniques, la majorité des patients ont été réinjectés entre 12 et 16 semaines après la précédente injection. Cependant, chez certains patients l’effet clinique recherché a duré plus longtemps c’est-à-dire jusqu’à 20 semaines.
Au moment du renouvellement de l’injection, l’évaluation du degré et du type de spasticité peut conduire à une modification de la dose et du choix des muscles injectés.
L’amélioration clinique devrait être observée 1 semaine après l’injection.
Spasticité affectant à la fois les membres supérieurs et inférieurs
En cas de traitement de la spasticité affectant à la fois les membres supérieurs et les membres inférieurs au cours de la même séance d’injection, la dose de DYSPORT à injecter dans chaque membre doit être adaptée à chaque patient, sans dépasser la dose maximale totale de 1500 unités.
Chez le sujet âgé (≥ 65 ans)
Les personnes âgées doivent faire l’objet d’une surveillance pour évaluer leur tolérance au traitement compte tenu de la plus grande fréquence de maladie concomitante et de traitement associé chez ces patients.
Les données cliniques n’ont pas mis en évidence de différences de réponse entre les personnes âgées et les patients adultes plus jeunes.
Mode d’administration
A l’aide d’une aiguille stérile, introduire dans le flacon 1 ml, 2,5 ml ou 5 ml (fonction du volume du muscle cible) d’une solution injectable de chlorure de sodium à 0,9 pour cent (voir rubrique 6.6). On obtient ainsi une solution limpide contenant 500, 200 ou 100 UNITES SPEYWOOD/ml de substance active.
Utiliser une seringue de 1 ml ou de 5 ml en fonction du volume total à injecter.
Le produit doit être injecté par voie intra musculaire.
Bien que la localisation des sites d’injection puisse être déterminée par la palpation manuelle, il est recommandé de recourir à des techniques de guidage électromyographique ou de stimulation nerveuse ou d’échographie pour localiser les muscles concernés.
Toutefois en raison de la difficulté technique de ces méthodes de guidage, l’injection du jambier postérieur nécessite une formation et une compétence particulière du médecin injecteur.
L’amélioration clinique survient généralement au cours des deux semaines qui suivent la séance d’injection.
Les séances d’injections pourront être répétées si besoin pour maintenir l’effet clinique recherché, mais seront toujours espacées d’au moins 12 semaines.
INCONTINENCE URINAIRE DUE A UNE HYPERACTIVITE NEUROLOGIQUE DU DETRUSOR
Posologie
La dose recommandée est de 600 U. Une dose de 800 U peut être utilisée en cas de réponse insuffisante, ou chez les patients avec une forme sévère de la maladie (par exemple, selon la gravité des signes et symptômes et/ou des paramètres urodynamiques).
Dysport doit être administré aux patients qui effectuent régulièrement un sondage intermittent propre.
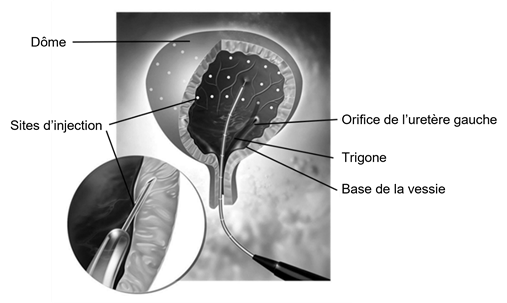
La dose totale administrée doit être répartie en 30 injections intra-détrusoriennes uniformément distribuées dans tout le détrusor, en évitant le trigone. Dysport est injecté à l’aide d’un cystoscope flexible ou rigide et chaque injection doit se faire à une profondeur d'environ 2 mm avec l'administration de 0,5 mL à chaque site. Pour la dernière injection, environ 0,5 mL de solution injectable de chlorure de sodium à 0,9 % (0,9 mg/mL) doit être injecté pour assurer une administration complète de la dose.
|
|
Une antibiothérapie prophylactique doit être initiée conformément aux recommandations et protocoles locaux ou conformément aux études cliniques (voir rubrique 5.1).
Les médicaments anticoagulants doivent être arrêtés au moins 3 jours avant l'administration de Dysport et repris uniquement le lendemain des injections. Si nécessaire, les héparines de bas poids moléculaire peuvent être administrées 24 heures avant l'administration de Dysport.
Avant l'injection, une anesthésie locale de l'urètre ou un gel lubrifiant peuvent être utilisés pour faciliter l'insertion du cystoscope. Si nécessaire, une instillation intravésicale d'une solution anesthésique diluée (avec ou sans sédation) ou une anesthésie générale peuvent également être effectuées. Si une instillation locale d'anesthésique est réalisée, la solution anesthésique devra être drainée, puis la vessie instillée (rincée) avec une solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) et drainée à nouveau avant de poursuivre la procédure d'injection intra-détrusorienne.
Avant l'injection, un volume suffisant de solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) doit être instillé dans la vessie afin d’obtenir une visualisation adéquate pour les injections.
Après l'administration des 30 injections intra-détrusoriennes, la solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) utilisée pour la visualisation de la paroi vésicale doit être drainée. Le patient doit rester sous observation pendant au moins 30 minutes après l'injection.
Le début de l'effet est généralement observé dans les 2 semaines suivant le traitement. Le traitement par Dysport doit être répété lorsque l'effet de l’injection précédente a diminué, mais pas plus tôt que 12 semaines après l'injection précédente. Dans les études cliniques (voir rubrique 5.1), le délai médian jusqu'au retraitement chez les patients traités par Dysport était compris entre 39 et 47 semaines. Toutefois, la durée de réponse peut être plus longue car plus de 40 % des patients n'avaient pas été retraités à 48 semaines.
Population pédiatrique
La sécurité et l'efficacité de Dysport pour le traitement de l'incontinence urinaire due à une hyperactivité neurologique du détrusor chez les enfants (moins de 18 ans) n'ont pas été établies.
Mode d'administration
Dysport est administré par injection intra-détrusorienne comme indiqué ci-dessus.
Lors du traitement de l'incontinence urinaire due à une hyperactivité neurologique du détrusor, Dysport est reconstitué avec une solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) pour donner une solution de 15 mL contenant soit 600 unités, soit 800 unités. Pour des instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
TRAITEMENT SYMPTOMATIQUE LOCAL DE LA SPASTICITÉ DES MEMBRES SUPERIEURS ET/OU INFÉRIEURS CHEZ L’ENFANT A PARTIR DE 2 ANS
Posologie
La dose à l’initiation ou lors d’une nouvelle séance d’injection doit être adaptée à chaque patient en fonction de la taille, du nombre et de la localisation des muscles concernés, de la sévérité de la spasticité, de l’existence d’une faiblesse musculaire localisée, de la réponse du patient au traitement précédent et/ou d’antécédents d’évènements indésirables avec la toxine botulinique.
Spasticité du membre inférieur chez l’enfant à partir de 2 ans
La dose totale maximale de DYSPORT administrée par séance d’injection ne doit pas dépasser 15 unités/kg en cas d’injections unilatérales ou 30 unités/kg en cas d’injections bilatérales. De plus, la dose totale de DYSPORT par séance d’injection ne doit pas dépasser la dose la plus faible entre les deux doses suivantes : 1 000 unités ou 30 unités/kg. La dose totale doit être répartie entre les différents muscles spastiques des membres inférieurs. Dans la mesure du possible, la dose doit être répartie sur plus d’un site d’injection par muscle. Le volume injecté ne doit pas dépasser 0,5 ml par site d’injection. Voir le tableau ci-dessous pour les doses recommandées par muscle.
|
Muscle |
Dose par muscle et par jambe (Unités Speywood par kg de poids corporel) |
Nombre de site d’injection par muscle |
|
Distal |
||
|
Gastrocnemius (gastrocnémien) |
5 à 15 U/kg |
Jusqu’à 4 |
|
Soleus (soléaire) |
4 à 6 U/kg |
Jusqu’à 2 |
|
Tibialis posterior (tibial postérieur) |
3 à 5 U/kg |
Jusqu’à 2 |
|
Proximal |
||
|
Biceps femoris, semitendinosus, semimembranosus (ischio jambiers) |
5 à 6 U/kg |
Jusqu’à 2 |
|
Sartorius (sartorius) |
3 à 15 U/kg |
Jusqu’à 2 |
|
Adductor magnus/longus/brevis (adducteurs) |
3 à 10 U/kg |
Jusqu’à 2 |
|
Rectus femoris, vastus medialis, vastus intermedius, vastus lateris (quadriceps) |
3 à 15 U/kg |
Jusqu’à 2 |
|
Dose totale |
Jusqu’à 15 U/kg/jambe si l’injection concerne uniquement les muscles distaux, uniquement les muscles proximaux ou multiniveaux (muscles proximaux et distaux). Ne doit pas dépasser la dose la plus faible entre les deux doses suivantes par séance d’injection : 1 000 unités ou 30 unités/kg. |
|
Le renouvellement de l’injection doit être réalisé lorsque l’effet clinique lié à la précédente injection diminue mais en respectant un intervalle minimum de 12 semaines après la précédente injection.
Dans les études cliniques, la majorité des patients ont été réinjectés entre 16 et 22 semaines après la précédente injection. Cependant, chez certains patients l’effet clinique recherché a duré plus longtemps par exemple jusqu’à 28 semaines.
Au moment du renouvellement de l’injection, l’évaluation du degré et du type de spasticité peut conduire à une modification de la dose et du choix des muscles injectés.
La posologie devra être réduite chez les enfants :
· présentant des comorbidités associées notamment troubles pré-existants de la déglutition ou respiratoires,
· dont les muscles à traiter sont peu développés,
· qui nécessitent une injection multisite,
· qui bénéficient d’injections sous anesthésie générale.
Dans tous les cas, lors du choix de la dose, une évaluation individuelle du rapport bénéfice/risque devra être envisagée, afin de réduire le risque d’effets indésirables notamment le risque de diffusion de la toxine à distance du site d’administration (voir rubriques 4.4 et 4.8).
Spasticité du membre supérieur chez l’enfant à partir de 2 ans
En cas d’injection unilatérale, la dose maximale de DYSPORT administrée par séance ne doit pas dépasser la plus faible des doses suivantes : 16 U/kg ou 640 U.
En cas d’injection bilatérale, la dose maximale de DYSPORT par séance ne doit pas dépasser la plus faible des doses suivantes : 21 U/kg ou 840 U.
La dose totale doit être répartie entre les différents muscles spastiques du ou des membres supérieurs.
Le volume injecté ne doit pas dépasser 0,5 ml par site d’injection.
Voir le tableau ci-dessous pour les doses recommandées par muscle.
Dose de DYSPORT par muscle dans le traitement de la spasticité des membres supérieurs chez l’enfant
|
Muscle |
Dose recommandée par muscle du membre supérieur (en U/kg de poids corporel) |
Nombre de site d’injection par muscle |
|
Brachialis (brachial) |
3 à 6 U/kg |
Jusqu’à 2 |
|
Brachioradialis (brachio-radial) |
1,5 à 3 U/kg |
1 |
|
Biceps brachii (biceps brachial) |
3 à 6 U/kg |
Jusqu’à 2 |
|
Pronator Teres (rond pronateur) |
1 à 2 U/kg |
1 |
|
Pronator quadratus (muscle carré pronateur) |
0,5 à 1 U/kg |
1 |
|
Flexor carpi radialis (fléchisseur radial du carpe) |
2 à 4 U/kg |
Jusqu’à 2 |
|
Flexor carpi ulnaris (fléchisseur ulnaire du carpe) |
1,5 à 3 U/kg |
1 |
|
Flexor digitorum profundus (fléchisseur profond des doigts) |
1 à 2 U/kg |
1 |
|
Flexor digitorum superficialis (fléchisseur superficiel des doigts) |
1,5 à 3 U/kg |
Jusqu’à 4 |
|
Flexor pollicis brevis/opponens pollicis (court fléchisseur du pouce/muscle opposant du pouce) |
0,5 à 1 U/kg |
1 |
|
Adductor Pollicis (adducteur du pouce) |
0,5 à 1 U/kg |
1 |
|
Dose totale |
Jusqu’à 16 U/kg pour chaque membre supérieur (sans dépasser 640 U par membre) Jusqu’à 21 U/kg en cas d’injection dans les deux membres supérieurs (sans dépasser 840 U) |
|
Le traitement par DYSPORT doit être renouvelé lorsque l’effet de la précédente injection a diminué, mais en respectant un intervalle minimum de 16 semaines après la précédente injection. Dans l’étude clinique, la majorité des patients ont été réinjectés entre 16 et 28 semaines après la précédente injection. Cependant, certains patients ont eu une réponse plus longue, à savoir 34 semaines ou plus. Au moment du renouvellement de l’injection, l’évaluation du degré et du type de spasticité peut conduire à une modification de la dose et du choix des muscles injectés.
Spasticité des membres supérieurs et inférieurs chez l’enfant à partir de 2 ans
En cas de traitement de la spasticité affectant à la fois les membres supérieurs et les membres inférieurs au cours de la même séance d’injection, la dose de DYSPORT à injecter dans chaque membre doit être adaptée à chaque patient et définie conformément à la rubrique posologie des membres correspondants (membres supérieurs et membres inférieurs), sans dépasser la dose maximale totale la plus faible entre les deux doses suivantes de 30 U/kg ou 1000 U par séance d’injection.
Le traitement ne doit pas être renouvelé avant 12 à 16 semaines après la précédente séance d’injection. Le délai avant le renouvellement du traitement sera défini pour chaque patient en fonction de son évolution et de la réponse au traitement.
Mode d’administration
DYSPORT est reconstitué avec une solution injectable de chlorure de sodium à 0,9 % (voir rubrique 6.6) et injecté par voie intramusculaire comme indiqué ci-dessus.
Bien que la localisation des sites d’injection puisse être déterminée par la palpation manuelle, il est recommandé de recourir à des techniques de guidage électromyographique ou de stimulation nerveuse ou d’échographie pour localiser les muscles concernés.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Infection urinaire au moment du traitement dans le cas de la prise en charge de l'incontinence urinaire due à une hyperactivité neurologique du détrusor.
4.4. Mises en garde spéciales et précautions d'emploi
La concentration de la solution reconstituée de DYSPORT est exprimée en Unités Speywood.
Étant donné l’absence d’harmonisation des systèmes d’unités pour les différentes toxines botuliniques commercialisées, il est nécessaire de faire preuve d’une extrême prudence au cas où le passage d’une toxine botulinique d’un laboratoire pharmaceutique à la toxine botulinique d’un autre laboratoire pharmaceutique s’avèrerait nécessaire.
Des effets indésirables, liés à la diffusion de la toxine à distance du site d’administration, ont été rapportés (cf. 4.8 Effets indésirables). Les patients traités à dose thérapeutique peuvent présenter une faiblesse musculaire excessive. Le risque de survenue de tels effets indésirables peut être réduit en ayant recours à la plus faible dose efficace et en ne dépassant pas la dose maximale recommandée.
De très rares cas de décès faisant parfois suite à une dysphagie, une pneumopathie (incluant de façon non limitative : dyspnée, insuffisance respiratoire, arrêt respiratoire) et/ou chez des patients ayant une asthénie significative ont été rapportés après traitement par la toxine botulinique de type A ou B.
DYSPORT doit être administré avec précaution chez les patients présentant des antécédents de troubles de la déglutition ou de troubles respiratoires car la diffusion de la toxine dans les muscles impliqués peut aggraver ces troubles. Une pneumopathie d’inhalation a été observée dans de rares cas et représente un risque chez les patients présentant des troubles respiratoires chroniques.
DYSPORT doit être utilisé uniquement avec prudence et sous étroite surveillance médicale chez les patients présentant un déficit marqué de la transmission neuromusculaire (exemple : myasthénie grave) clinique ou infraclinique. Ces patients peuvent présenter une sensibilité accrue aux produits tels que DYSPORT pouvant conduire à une faiblesse excessive des muscles.
Les patients et leur entourage doivent être avertis de la nécessité d’une prise en charge médicale immédiate en cas de troubles de la déglutition, de troubles du langage ou de troubles respiratoires.
La posologie et la fréquence d’administration recommandées ne doivent pas être dépassées.
DYSPORT ne doit pas être administré pour traiter la spasticité d’un patient présentant une contracture fixe.
La prudence est de rigueur lors du traitement de patients adultes, en particulier les sujets âgés, présentant une spasticité des membres inférieurs, car ils peuvent présenter un risque plus élevé de chute. Au cours des études cliniques contrôlées par placebo dans le traitement de la spasticité des membres inférieurs, des cas de chute ont été rapportés chez 6,3% et 3,7% des patients dans les groupes DYSPORT et placebo respectivement.
L’existence d’antécédents d’atteinte neurogène de la face (paralysie faciale, polyradiculonévrite) nécessite, lors de la première injection, d’utiliser des doses égales au quart de la dose recommandée (cf. 4.2 Posologie et mode d’administration).
Les patients souffrant de blépharospasme peuvent avoir été sédentaires pendant un très long moment. En conséquence, lors d’un traitement par la toxine botulinique, il est nécessaire de leur conseiller une reprise d’activité progressive.
La diminution du clignement due à l’injection de la toxine botulinique dans le muscle orbiculaire peut conduire à une exposition prolongée de la cornée, à une lésion épithéliale persistante et à une ulcération de la cornée en particulier chez les patients ayant présenté une paralysie faciale. Dans ce cas, des mesures préventives et curatives doivent être prises.
Des cas de sécheresse oculaire ont été rapportés lors de l'utilisation de DYSPORT dans les régions périoculaires (voir rubrique 4.8). Il est important de porter une attention particulière à cet effet indésirable car la sécheresse oculaire peut prédisposer à des troubles cornéens. Des gouttes protectrices, des pommades, l’occlusion de l'œil par un pansement ou d'autres moyens peuvent être nécessaires pour prévenir les troubles cornéens.
Comme pour toute injection intramusculaire, DYSPORT ne doit être injecté que si strictement nécessaire, chez les patients présentant des temps de saignement allongés ou une infection/inflammation au niveau du site d’injection.
En cas d’atrophie du muscle ciblé, des précautions particulières doivent être prises. Des cas d'atrophie musculaire ont été rapportés après l’utilisation de toxine botulinique (voir rubrique 4.8).
DYSPORT ne doit être utilisé que pour le traitement d’un seul patient, au cours d’une même séance d’injection.
Toute fraction de solution restante doit être éliminée conformément aux instructions pour l’élimination et la manipulation (cf. section 6.6).
Des précautions particulières doivent être prises pour la préparation et l’administration du produit, ainsi que pour l’inactivation et l’élimination de la solution reconstituée non utilisée (voire rubrique 6.6).
Ce produit contient une faible quantité d’albumine humaine. Le risque de transmission d’une infection virale ne peut être totalement exclu après utilisation de sang humain ou de dérivés sanguins.
La formation d’anticorps dirigés contre la toxine botulinique n’a été que rarement observée chez les patients traités par DYSPORT. Au plan clinique, la présence d’anticorps neutralisants est suspectée en cas de diminution de la réponse au traitement et/ou de la nécessité d’augmenter constamment les doses.
Population pédiatrique
Dans la déformation dynamique du pied en équin chez les enfants présentant une paralysie cérébrale, une évaluation fonctionnelle initiale précise doit être effectuée en milieu spécialisé. Elle permet :
· d’évaluer la pertinence de l’indication :
o spasticité prédominante,
o absence de faiblesse musculaire parfois masquée par l’hypertonie. Cette faiblesse pourrait être aggravée par une injection de toxine botulinique,
o absence de rétraction fixée importante ou de cicatrice post chirurgicale rendant inutile une injection de toxine botulinique.
· de déterminer les différentes composantes du traitement (kinésithérapie, port d’attelles…),
· d’adapter le traitement en fonction de l’évolution clinique.
Pour le traitement de la spasticité associée à une paralysie cérébrale chez les enfants, DYSPORT ne doit être utilisé que chez les enfants de 2 ans ou plus. Après la commercialisation, des signalements concernant une éventuelle diffusion de la toxine à distance du site d’injection ont été très rarement rapportés chez les patients pédiatriques présentant des comorbidités, principalement en cas de paralysie cérébrale. En général, la dose utilisée dans ces cas était supérieure à celle recommandée (voir rubrique 4.8).
De rare signalements spontanés de décès, parfois associés à une pneumonie d’inhalation, ont été rapportés chez des enfants atteints de paralysie cérébrale grave après un traitement par la toxine botulinique, y compris après une utilisation non conforme à l’autorisation de mise sur le marché (par exemple au niveau du cou). Le traitement des patients pédiatriques présentant des troubles neurologiques graves, une dysphagie ou ayant des antécédents de pneumonie d’inhalation ou de maladie des poumons nécessite une extrême prudence. Chez les patients présentant un mauvais état général, le traitement doit être administré uniquement si le bénéfice potentiel pour le patient est supérieur aux risques.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L’effet de la toxine botulinique peut être potentialisé par d’autres médicaments (aminoglycosides, curares, anticholinestérasiques, etc.) interférant directement ou indirectement sur la transmission neuromusculaire, l’utilisation de tels médicaments doit se faire avec prudence chez les patients traités par toxine botulinique.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études réalisées chez l’animal n’ont pas mis en évidence d’effet délétère direct ou indirect sur la grossesse, le développement fœtal/embryonnaire, la parturition ou le développement post natal, excepté à des doses très élevées, toxiques pour la mère (cf. section 5.3). Les données relatives à l’utilisation du complexe toxine botulinique de type A-hémagglutinine chez la femme enceinte sont limitées.
DYSPORT ne doit pas être utilisé chez la femme enceinte sauf si le bénéfice attendu justifie le risque encouru par le fœtus.
Le passage dans le lait maternel du complexe toxine botulinique de type A - hémagglutinine n’est pas connu. L’utilisation du complexe toxine botulinique de type A - hémagglutinine n’est pas recommandée pendant l’allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
La fréquence des effets indésirables est classée comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Général
|
Classes de systèmes d’organes |
Fréquent |
Peu fréquent |
Rare |
|
Affections du système nerveux |
|
|
amyotrophie névralgique |
|
Affections de la peau et du tissu sous-cutané |
|
prurit |
rash |
|
Troubles généraux et anomalies au site d'administration |
asthénie |
|
|
|
fatigue |
|
|
|
|
syndrome pseudo-grippal |
|
|
|
|
douleur/hématome au point d’injection |
|
|
Blépharospasme et spasme hémifacial
Les effets indésirables suivants ont été observés chez les patients traités avec DYSPORT pour un blépharospasme ou un spasme hémifacial.
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
|
Affections du système nerveux |
|
parésie faciale |
paralysie du nerf facial (septième nerf crânien) |
|
|
Affections oculaires |
ptosis |
diplopie |
|
ophtalmoplégie |
|
|
sécheresse oculaire |
|
|
|
|
|
augmentation de la sécrétion lacrymale |
|
|
|
|
Affections de la peau et du tissu sous cutané |
|
œdème palpébral |
|
entropion |
Les effets indésirables peuvent survenir suite à une injection profonde ou mal localisée de DYSPORT entraînant une paralysie temporaire des muscles proches du site d’injection.
Torticolis spasmodique
Les effets indésirables suivants ont été observés chez des patients traités par DYSPORT pour un torticolis spasmodique.
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
|
Affections du système nerveux |
|
céphalées |
|
|
|
|
sensations vertigineuses |
|
|
|
|
|
parésie faciale |
|
|
|
|
Affections oculaires |
|
vision trouble |
diplopie |
|
|
|
baisse de l’acuité visuelle |
ptosis |
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
dysphonie |
|
aspirations du contenu gastro-intestinal dans les voies respiratoires (fausse route) |
|
|
dyspnée |
|
|
|
|
Affections gastro-intestinales |
dysphagie |
|
|
|
|
sécheresse de la bouche |
|
nausées |
|
|
|
Affections musculo-squelettiques et systémiques |
faiblesse musculaire |
douleur cervicale |
atrophie musculaire |
|
|
|
douleur musculo-squelettique |
atteinte de la mâchoire |
|
|
|
|
myalgie |
|
|
|
|
|
douleurs distales |
|
|
|
|
|
raideur musculo-squelettique |
|
|
Les dysphagies sont a priori dose dépendantes et surviennent le plus souvent après une injection dans le muscle sterno-cléido-mastoïdien. Un régime alimentaire léger doit être mis en place en attendant la résolution des symptômes.
Traitement symptomatique local de la spasticité affectant les membres supérieurs et/ou inférieurs chez l’adulte
Spasticité des membres inférieurs chez l’adulte
Les effets indésirables suivants ont été observés chez des patients adultes traités par DYSPORT pour une spasticité affectant les membres inférieurs.
|
Classes de systèmes d’organes |
Fréquent |
|
Affections gastro-intestinales |
dysphagie |
|
Affections musculo-squelettiques et systémiques |
faiblesse musculaire, myalgie |
|
Troubles généraux et anomalies au site d'administration |
asthénie |
|
fatigue |
|
|
syndrome pseudo-grippal |
|
|
réaction au site d’injection (douleur, hématomes, rash, prurit) |
|
|
Lésions, intoxications et complications liées aux procédures |
chute |
Spasticité des membres supérieurs chez l’adulte
Les effets indésirables suivants ont été observés chez des patients adultes traités par DYSPORT pour une spasticité affectant les membres supérieurs.
|
Fréquent |
Peu fréquent |
|
|
Affections gastro-intestinales |
|
dysphagie* |
|
Affections musculo-squelettiques et systémiques |
faiblesse musculaire du membre supérieur |
|
|
douleur musculo-squelettique du membre supérieur |
|
|
|
douleur aux extrémités |
|
|
|
Troubles généraux et anomalies au site d'administration |
réactions au point d’injection (telles que douleur, érythème, gonflement…) |
|
|
asthénie |
|
|
|
fatigue |
|
|
|
syndrome pseudo-grippal |
|
* La fréquence relative aux dysphagies est issue de l’analyse combinée des essais cliniques réalisés en ouvert. Aucun cas de dysphagie n’a été rapporté dans les études cliniques conduites en double aveugle.
Spasticité des membres supérieurs et inférieurs chez l’adulte
Les effets indésirables suivants ont été observés chez des patients adultes traités par DYSPORT pour une spasticité affectant à la fois les membres inférieurs et les membres supérieurs.
|
Classes de systèmes d’organes |
Fréquent |
Peu fréquent |
|
Affections gastro-intestinales |
dysphagie |
|
|
Affections musculo-squelettiques et systémiques |
faiblesse musculaire du membre inférieur |
|
|
faiblesse musculaire du membre supérieur |
|
|
|
Affections du rein et des voies urinaires |
|
incontinence urinaire |
|
Troubles généraux et anomalies au site d'administration |
troubles de la démarche |
|
|
Lésions, intoxications et complications liées aux procédures |
blessure accidentelle |
|
|
chute |
|
Incontinence urinaire due à une hyperactivité neurologique du détrusor
|
Classes de systèmes d’organes |
Fréquent |
Peu fréquent |
|
Infections et infestations |
Infection des voies urinairesa,b |
|
|
Bactériuriea |
||
|
Affections du système nerveux |
Céphalée |
Hypoesthésie |
|
Affections gastro-intestinales |
Constipation |
|
|
Affections musculo-squelettiques et systémiques |
|
Faiblesse musculaire |
|
Affections du rein et des voies urinaires |
Hématuriea |
Rétention urinairec |
|
Hémorragie uréthrale |
||
|
Hémorragie de la vessie |
||
|
Affections des organes de reproduction et du sein |
Dysérection |
|
|
Troubles généraux et anomalies au site d'administration |
Fièvre |
Vessie douloureusea |
|
Lésions, intoxications et complications liées aux procédures |
|
Dysréflexie autonome |
a Possiblement lié à l’intervention
b Dans les études pivots en double aveugle contrôlées versus placebo, au cours des 2 premières semaines suivant le traitement, des infections des voies urinaires ont été signalées chez 4,0 % des patients traités par Dysport et 6,2 % des patients sous placebo. Les infections des voies urinaires peuvent induire des pyélonéphrites
c Peut survenir si les patients n’ont pas un calendrier de sondage adapté.
Traitement symptomatique local de la spasticité des membres supérieurs et/ou inférieurs chez l’enfant à partir de 2 ans
Spasticité des membres inférieurs chez l’enfant
Les effets indésirables suivants ont été observés chez les enfants traités par DYSPORT pour une spasticité affectant les muscles des membres inférieurs.
|
Classes de systèmes d’organes |
Fréquent |
Peu fréquent |
|
Affections musculo-squelettiques et systémiques |
Myalgie |
|
|
Faiblesse musculaire |
||
|
Affections du rein et des voies urinaires |
Incontinence urinaire |
|
|
Troubles généraux et anomalies au site d'administration |
Syndrome grippal |
Asthénie |
|
Réaction au site d’injection (tels que douleur, érythème, bleus…) |
||
|
Troubles de la démarche |
||
|
Fatigue |
||
|
Lésions, intoxications et complications liées aux procédures |
Chute |
|
Spasticité des membres supérieurs chez l’enfant
Les effets indésirables suivants ont été observés chez les enfants traités par DYSPORT pour une spasticité affectant les muscles des membres supérieurs.
|
Classes de systèmes d’organes |
Fréquent |
Peu fréquent |
|
Affections musculo-squelettiques et systémiques |
Faiblesse musculaire |
Myalgie |
|
Extrémités douloureuses |
||
|
Troubles généraux et anomalies au site d'administration |
Syndrome grippal |
|
|
Asthénie |
|
|
|
Fatigue |
|
|
|
Réactions au site d’injection (telles que douleur, rash, contusion, gonflement, eczéma…) |
|
|
|
Lésions, intoxications et complications liées aux procédures |
Rash |
|
Spasticité des membres supérieurs et inférieurs chez l’enfant
Lors du traitement concomitant des membres supérieurs et inférieurs avec DYSPORT à une dose totale ne dépassant la plus faible des doses suivantes, 30 U/kg ou 1 000 U, il n'y a pas de nouvelles données de sécurité par rapport à celles attendues pour le traitement séparé des muscles des membres supérieurs ou des muscles des membres inférieurs.
Les chutes et les troubles de la démarche peuvent être dus à une faiblesse excessive du muscle traité et/ou à une diffusion locale de DYSPORT au niveau des autres muscles impliqués dans la marche et l’équilibre.
Expérience post commercialisation
Le profil d’effets indésirables rapportés au laboratoire après commercialisation est cohérent avec la pharmacologie du produit et reflète le profil observé pendant les études cliniques.
|
Classes de systèmes d’organes |
Fréquence indéterminée |
|
Affections du système immunitaire |
Hypersensibilité |
|
Affections du système nerveux |
Hypoesthésie |
|
Affections musculo-squelettiques et systémiques |
Atrophie musculaire |
Les effets indésirables résultant de la diffusion de la toxine à distance du site d’injection ont été très rarement observés (faiblesse musculaire excessive, dysphagie, pneumopathie d’inhalation qui peuvent être fatales) (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Des doses excessives peuvent provoquer à distance de profondes paralysies neuromusculaires.
Un surdosage peut augmenter le risque que la neurotoxine pénètre dans la circulation sanguine et peut conduire à des complications associées au syndrome botulinique (ex dysphagie et dysphonie). Une réanimation respiratoire peut être nécessaire si des doses excessives provoquent une paralysie des muscles respiratoires. Des soins médicaux généraux doivent être conseillés.
Dans le cas d’un surdosage, le patient doit être placé sous surveillance médicale pour rechercher d’éventuels signes et /ou symptômes de faiblesse musculaire excessive ou de paralysie musculaire.
Un traitement symptomatique doit être mis en place si nécessaire.
Les symptômes de surdosage ne se manifestent pas obligatoirement juste après l’injection. En cas d’injection ou d’ingestion orale accidentelle, le patient doit être placé sous surveillance médicale pendant plusieurs semaines afin de rechercher d’éventuels signes et/ou symptômes de faiblesse musculaire excessive ou de paralysie musculaire.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Autres myorelaxants à action périphérique, code ATC : M03AX01.
La toxine botulinique A est produite par Clostridium botulinum.
Elle bloque la libération d’acétylcholine dans la jonction neuro-musculaire, entraînant une dégénérescence des terminaisons nerveuses et donc une paralysie.
Ce blocage est définitif, mais l’existence de phénomènes de repousse axonale explique le caractère réversible des paralysies induites par les injections de toxine.
Efficacité et sécurité clinique
Traitement symptomatique de la spasticité des membres supérieurs chez l’adulte
L’efficacité et la sécurité de DYSPORT dans le traitement de la spasticité des membres supérieurs chez l’adulte ont été évaluées dans le cadre d’une étude multicentrique, randomisée, en double aveugle, comparativement à un placebo, incluant 238 patients (159 traités par DYSPORT et 79 traités par placebo) souffrant de spasticité des membres supérieurs, soit après un accident vasculaire cérébral (90%) ou un traumatisme cérébral (10%) survenus au moins six mois auparavant.
Le Principal Groupe Musculaire Cible (PGMC) était les muscles fléchisseurs des doigts (56%) suivis des fléchisseurs du coude (28%) et des fléchisseurs du poignet (16%).
Le critère principal d’évaluation de l’efficacité était le tonus musculaire du PGMC à la semaine 4, mesuré au moyen de l’Echelle d’Ashworth Modifiée (MAS). Les principaux résultats obtenus aux semaines 4 et 12 sont présentés ci-dessous :
|
|
Semaine 4 |
Semaine 12 |
||||
|
Placebo |
DYSPORT 500 U |
DYSPORT 1 000 U |
Placebo |
DYSPORT 500 U |
DYSPORT 1 000 U |
|
|
n=79 |
n=80 |
n=79 |
n=79 |
n=80 |
n=79 |
|
|
Tonus musculaire sur la MAS : variation moyenne (MC) depuis l’inclusion sur le PGMC |
-0,3 |
-1,2** |
-1,4** |
-0,1 |
-0,7** |
-0,8** |
|
|
|
|
n=75 |
n=76 |
n=76 |
|
|
Tonus musculaire sur la MAS : variation moyenne (MC) depuis l’inclusion sur les fléchisseurs du poignet |
-0,3 |
-1,4** |
-1,6** |
-0,3 |
-0,7* |
-0,9* |
|
n=54 |
n=57 |
n=58 |
n=52 |
n=54 |
n=56 |
|
|
Tonus musculaire sur la MAS : variation moyenne (MC) depuis l’inclusion sur les fléchisseurs des doigts |
-0,3 |
-0,9* |
-1,2** |
-0,1 |
-0,4* |
-0,6* |
|
n=70 |
n=66 |
n=73 |
n=67 |
n=62 |
n=70 |
|
|
Tonus musculaire sur la MAS : variation moyenne (MC) depuis l’inclusion sur les fléchisseurs du coude |
-0,3 |
-1,0* |
-1,2** |
-0,3 |
-0,7* |
-0,8* |
|
n=56 |
n=61 |
n=48 |
n=53 |
n=58 |
n=46 |
|
|
Tonus musculaire sur la MAS : variation moyenne (MC) depuis l’inclusion sur les extenseurs de l’épaule (1) |
-0,4 |
-0,6 |
-0,7 |
0,0 |
-0,9 |
0,0 |
|
n=12 |
n=7 |
n=6 |
n=12 |
n=7 |
n=6 |
|
|
*p<0,05 **p<0,0001 MC = moindres carrés (1) Aucun test statistique n’a été effectué en raison de la fréquence réduite par groupe de traitement et groupe placebo |
||||||
Dans une étude de suivi, ouverte, d’une durée de 15 mois, le renouvellement de l’injection a été décidé en fonction des besoins cliniques après un minimum de 12 semaines. L’objectif principal de cette étude de suivi était d’évaluer la sécurité à long terme de DYSPORT. Les effets indésirables sous traitement observés étaient généralement d’intensité légère à modérée. Par ailleurs, l’évaluation de critères secondaires (exploratoires) semble suggérer que l'efficacité de DYSPORT est maintenue jusqu'à 1 an d’après l’échelle MAS (Modified Ashworth Scale) et l’échelle de Tardieu.
Traitement symptomatique de la spasticité des membres inférieurs chez l’adulte
L'efficacité et la sécurité de DYSPORT dans le traitement de la spasticité des membres inférieurs ont été évalués dans le cadre d’une étude pivot multicentrique randomisée, en double-aveugle, contrôlée par placebo, qui a inclus 385 patients (255 traités par DYSPORT et 130 traités par placebo) souffrant de spasticité des membres inférieurs après un accident vasculaire cérébral ou un traumatisme cérébral. Le critère principal était le score de l’Echelle d’Ashworth Modifiée (MAS) évalué au niveau de l'articulation de la cheville.
Un volume total de 7,5 ml de DYSPORT 1000 unités (N = 125), de DYSPORT 1500 unités (N = 128) ou de placebo (N = 128) a été réparti entre les muscles gastrocnémiens et soléaires et au moins un autre muscle du membre inférieur en fonction du tableau clinique.
Une amélioration statistiquement significative du score de MAS a été rapportée à la dose de 1500 unités lors de l’évaluation à la cheville avec le genou en extension (impliquant tous les fléchisseurs plantaires). Une amélioration statistiquement significative du score de MAS a été rapportée aux doses de 1000 et 1500 unités lors de l'évaluation à la cheville avec le genou fléchi (impliquant tous les fléchisseurs plantaires à l'exception du gastrocnémien).
Une amélioration de la spasticité évaluée au niveau de l'articulation de la cheville au moyen de l'Echelle de Tardieu (TS) a également été rapportée, avec des améliorations statistiquement significatives du grade de sévérité de la spasticité aux doses de 1000 et 1500 unités. Le traitement par DYSPORT a également été associé à une amélioration clinique, telle que mesurée par l’échelle d’évaluation globale par le médecin (Physician Global Assessment, PGA), statistiquement significative aux deux doses.
À l'issue de cette étude, 345 patients ont été inclus dans une étude d'extension en ouvert au cours de laquelle un renouvellement du traitement par DYSPORT aux doses de 1000 ou 1500 unités était envisagé selon les besoins. Les patients présentant également une spasticité des membres supérieurs ont pu recevoir des injections de DYSPORT à la dose de 500 unités dans le membre supérieur atteint, en plus des 1000 unités dans le membre inférieur, sans dépasser une dose totale maximale de 1500 unités. L’amélioration des paramètres d'efficacité (MAS, PGA et TS) observée après 4 semaines de traitement par DYSPORT en double aveugle dans le membre inférieur s’est maintenue après des traitements répétés. Au cours de l'étude en double aveugle, aucune amélioration de la vitesse de la marche n'a été observée après un traitement unique, mais une amélioration a été observée après un traitement répété.
Une autre étude contrôlée en double aveugle a été menée dans le traitement de la spasticité des adducteurs de la hanche chez 74 sujets atteints de sclérose en plaques, recevant soit un placebo, soit DYSPORT 500, 1000 ou 1500 unités. Le médicament actif ou le placebo ont été répartis entre le grand adducteur, le petit adducteur et l’adducteur moyen des deux jambes. Une amélioration statistiquement significative de la distance entre les genoux a été rapportée dans le groupe DYSPORT 1500 unités par rapport au placebo.
Incontinence urinaire due à une hyperactivité neurologique du détrusor
Deux études cliniques pivots randomisées, en double aveugle, contrôlées versus placebo et multicentriques ont été menées chez des patients souffrant d'incontinence urinaire due à une hyperactivité neurologique du détrusor. Tous les patients pratiquaient déjà un sondage pour vider régulièrement leur vessie et étaient insuffisamment contrôlés par des traitements oraux ; les patients étaient naïfs ou non naïfs d’un traitement antérieur par la toxine botulinique pour voie intra-détrusorienne. Dans les deux études, un total de 485 patients avec une blessure médullaire (N = 341) ou de sclérose en plaques (N = 144) ont été randomisés pour recevoir Dysport 600 U (N = 162), Dysport 800 U (N = 161), ou le placebo (N = 162). Le traitement a été administré par cystoscopie en 30 injections intra-détrusoriennes uniformément réparties, en évitant le trigone. Une antibiothérapie prophylactique a été débutée au moins 3 jours avant l'administration de Dysport et poursuivie pendant au moins 3 jours après l'administration de Dysport. Après le traitement initial, les patients pouvaient recevoir d'autres injections de Dysport 600 U ou Dysport 800 U s'ils remplissaient les critères de retraitement.
Le critère principal d'évaluation de l'efficacité était la variation du nombre d’épisodes hebdomadaires d'incontinence urinaire entre l’inclusion et la semaine 6. Les critères d'évaluation secondaires comprenaient le pourcentage de patients sans épisodes d'incontinence urinaire (réduction de 100 %) à la semaine 6, la variation du volume par miction entre l’inclusion et la semaine 6, une série de paramètres urodynamiques (cystométrie de remplissage), un questionnaire de qualité de vie des patients spécifique de l'incontinence (I-QOL ; comprend les comportements d'évitement et de limitation, l'impact psychosocial et la gêne sociale) et l'impression globale de la réponse au traitement.
Les résultats combinés des études pivots sont présentés dans le tableau ci-dessous :
Critères d'évaluation primaires et secondaires dans les études pivots combinées (population randomisée)
|
|
Placebo (N = 162) |
Dysport 600 U (N = 162) |
Dysport 800 U (N = 161) |
|
Episodes hebdomadaires d'incontinence urinaire |
|
|
|
|
Semaine 2 |
|
|
|
|
Variation moyenne* (ET) |
-11,3 (1,4) |
-19,9 (1,4) |
-21,9 (1,4) |
|
Différence vs placebo (IC95 %) |
|
-8,6 (-12,2 ; -4,9) |
-10,6 (-14,3 ; -7,0) |
|
p |
|
<0,0001 |
<0,0001 |
|
Semaine 6 |
|
|
|
|
Variation moyenne* (ET) |
-12,7 (1,4) |
-22,7 (1,3) |
-23,6 (1,3) |
|
Différence vs placebo (95% CI) |
|
-10,0 (-13,5 ; -6,5) |
-10,9 (-14,4 ; -7,4) |
|
p |
|
<0,0001 |
<0,0001 |
|
Semaine 12 |
|
|
|
|
Variation moyenne* (ET) |
-9,2 (1,5) |
-20,4 (1,5) |
-22,8 (1,5) |
|
Différence vs placebo (IC95%) |
|
-11,3 (-15,2 ; -7,3) |
-13,6 (-17,6 ; -9,7) |
|
p |
|
<0,0001 |
<0,0001 |
|
Absence d’épisodes d’incontinence urinaire, Semaine 6 [a] |
|
|
|
|
Pourcentage de patients |
2,9% |
36,1% |
28,8% |
|
Odds ratio vs placebo (IC95%) |
|
18,9 (6,9 ; 51,9) |
15,5 (5,6 ; 42,9) |
|
p |
|
<0,0001 |
<0,0001 |
|
Capacité cystomanométrique maximale (mL), Semaine 6 [b] |
|
|
|
|
Variation moyenne (ET) |
-4,0 (13,9) |
164,6 (13,6) |
175,8 (13,7) |
|
Différence vs placebo (IC95%) |
|
168,5 (132,4 ; 204,7) |
179,8 (143,5 ; 216,1) |
|
p |
|
<0,0001 |
<0,0001 |
|
Absence de contractions involontaire du détrusor, Semaine 6 [b] |
|
|
|
|
Pourcentage de patients |
6,6% |
44,0% |
55,0% |
|
Odds ratio vs placebo (IC95%) |
|
11,9 (5,3 ; 26,6) |
18,6 (8,3 ; 41,7) |
|
p |
|
<0,0001 |
<0,0001 |
|
Volume lors de la première contraction involontaire du détrusor (mL), Semaine 6 [b] |
|
|
|
|
Variation moyenne (ET) |
12,3 (14,7) |
166,4 (14,4) |
191,2 (14,6) |
|
Différence vs placebo (IC95%) |
|
154,1 (116,0 ; 192,1) |
178,9 (140,4 ; 217,5) |
|
p |
|
<0,0001 |
<0,0001 |
|
Pression détrusorienne maximale pendant le stockage (cmH2O), Semaine 6 [b] |
|
|
|
|
Variation moyenne (ET) |
-4,9 (2,3) |
-33,1 (2,2) |
-35,4 (2,2) |
|
Différence vs placebo (IC95%) |
|
-28,2 (-34,0 ; -22,3) |
-30,4 (-36,3 ; -24,5) |
|
p |
|
<0,0001 |
<0,0001 |
|
Score I-QOL total [c], Semaine 6 |
|
|
|
|
Variation moyenne (ET) |
7,1 (1,8) |
22,1 (1,8) |
22,2 (1,7) |
|
Différence vs placebo (IC95%) |
|
15,0 (10,4 ; 19,6) |
15,1 (10,5 ; 19,7) |
|
p |
|
<0,0001 |
<0,0001 |
|
* estimée par la méthode des moindres carrés [a] Le pourcentage de patients pour lesquels la réduction des épisodes d’incontinence était d’au moins 75% de l’inclusion à la semaine 6 était de : 62,5 % et 57,6 % respectivement dans le groupe Dysport 600 U et 800 U versus 15,0 % dans le groupe placebo. Le pourcentage correspondant de patients avec une réduction d’au moins 50 % était de 73,6 % et 67,6 % versus 34,3 %. |
|||
Dans les deux groupes Dysport comparativement au groupe placebo, des améliorations significatives du volume par miction et du paramètre urodynamique de la compliance détrusorienne par rapport aux données à l’inclusion ont été observées. En plus de la qualité de vie liée à la santé spécifique de l'incontinence mesurée par I-QOL, l'impression globale du patient sur la réponse au traitement, telle que mesurée par l'échelle d'évaluation à 7 niveaux (de "beaucoup mieux" à "bien pire") a montré une réponse significativement meilleure après le traitement Dysport par rapport au placebo.
Pour tous les critères d'efficacité, les patients ont présenté une réponse similaire en cas de renouvellement de l’injection de Dysport. Quatre-cent-vingt-six (426), 217 et 76 patients ont reçu respectivement au moins 1, 2 et 3 administrations de Dysport. La diminution moyenne des épisodes hebdomadaires d'incontinence urinaire à la semaine 6 sur l'ensemble des cycles Dysport était de
-21,2 à -22,3 pour Dysport 600 U et de -21,3 à -23,7 pour Dysport 800 U.
Le délai médian de retraitement était de 39 à 47 semaines après le traitement initial par Dysport. Cependant plus de 40 % des patients n'avaient pas été retraités à 48 semaines.
Traitement symptomatique de la spasticité des membres inférieurs chez l’enfant à partir de 2 ans
Une étude multicentrique, en double aveugle, comparative versus placebo (étude Y-55-52120-141) a été réalisée chez des enfants atteints de paralysie cérébrale souffrant d’une déformation dynamique du pied en équin due à une spasticité. Au total, 235 patients (naïfs ou non d’un traitement par toxine botulinique) avec un tonus musculaire de grade 2 ou plus sur l’échelle modifiée d’Ashworth (Modified Ashworth Scale, MAS) ont été randomisés pour être traités soit par DYSPORT 10 U/kg/jambe soit par DYSPORT 15 U/kg/jambe soit par un placebo. Quarante et un pour cent (41%) des patients ont reçu des injections bilatérales correspondant à une dose totale de 20 U/kg ou 30 U/kg. Le critère d’évaluation principal était la variation moyenne sur l’échelle modifiée d’Ashworth (Modified Ashworth Scale, MAS) entre l’inclusion et la semaine 4, au niveau des fléchisseurs plantaires de la cheville.
Variation de la MAS entre le début de l’étude et la semaine 4 (population en ITT) :
|
Critère |
Placebo (n=77) |
DYSPORT |
|
|
|
|
10 U/kg/jambe (n=79) |
15 U/kg/jambe (n=79) |
|
Variation moyenne* de la MAS des fléchisseurs plantaires de la cheville entre le début de l’étude et la Semaine 4 |
-0,5 |
-0,9** |
-1,0*** |
|
*estimée par la méthode des moindres carrés **p≤0,003 par rapport au placebo ***p≤0,0006 par rapport au placebo |
|||
Une amélioration de la spasticité des fléchisseurs plantaires de la cheville a été observée sur l’échelle de Tardieu. DYSPORT a amélioré, de manière statistiquement significative, le grade de spasticité (Y) mesuré sur l’échelle de Tardieu, aux deux doses de 10 U/kg/jambe et 15 U/kg/jambe, par rapport au placebo, à la semaine 4. L’angle de ressaut (Xv3) était amélioré de manière statistiquement significative par rapport au placebo dans le groupe DYSPORT 15 U/kg/jambe à la semaine 4.
Les deux doses de DYSPORT (10 U/kg/jambe et 15 U/kg/jambe) ont démontré une amélioration significative du score moyen sur l’échelle de réalisation des objectifs (Goal Attainment Scale, GAS) à la semaine 4, Le score GAS mesurait la progression de la réalisation des objectifs de traitement sélectionnés au début de l'étude. Les 5 objectifs les plus souvent retenus étaient : une amélioration de la marche (70,2% des patients), une amélioration de l'équilibre (32,3%), une réduction de la fréquence des chutes (31,1%), une réduction des trébuchements (19,6%) et une amélioration de l'endurance (17,0%).
A l’issue de cette étude, 216 patients ont été inclus dans l’extension de l’étude, en phase ouverte (Y-55-55120-147), au cours de laquelle ils pouvaient recevoir une nouvelle injection de DYSPORT selon le besoin clinique. Dans cette étude, dont l’objectif principal était d’évaluer la sécurité à long terme de DYSPORT, les injections pouvaient être réalisées au niveau distal (gastrocnémien, soléaire et tibial postérieur) et proximal (ischio-jambiers et adducteurs de la hanche), et également aux deux niveaux.
Traitement symptomatique de la spasticité des membres supérieurs chez l’enfant à partir de 2 ans
L’efficacité et la sécurité de DYSPORT dans le traitement de la spasticité des membres supérieurs chez l’enfant à partir de 2 ans ont été évaluées dans une étude multicentrique, randomisée, en double aveugle, dans laquelle des doses de 8 U/kg et 16 U/kg ont été comparées à une dose faible de 2 U/kg administrée aux patients du groupe témoin (étude Y-52-52120-153). Un total de 210 patients naïfs ou non naïfs de toxine botulinique avec une spasticité des membres supérieurs due à une paralysie cérébrale a été randomisé et traité dans le cadre de l'étude.
Après le traitement initial, jusqu'à 3 autres séances d’injection de DYSPORT pouvaient être effectuées aux doses prévues de 8 U/kg ou de 16 U/kg, l'investigateur pouvant choisir d'augmenter ou de diminuer la dose (sans toutefois dépasser 16 U/kg).
La dose totale de DYSPORT a été injectée par voie intramusculaire dans les muscles des membres supérieurs concernés, ce qui comprenait le PGMC (principal groupe musculaire cible) parmi les fléchisseurs du coude ou des fléchisseurs du poignet ainsi que d'autres muscles du membre supérieur, selon la configuration de la spasticité.
Le volume maximal injecté par site d'injection était de 0,5 ml. Cependant, plusieurs sites d'injection par muscle étaient autorisés. Un repérage des muscles par stimulation électrique et/ou par échographie ont été utilisées pour faciliter la localisation musculaire pour l'injection.
Le critère principal d’efficacité était le changement moyen du score MAS (Modified Ashworth Scale) dans le PGMC entre sa valeur à l’inclusion et à la semaine 6. Les critères d'efficacité secondaires étaient le score moyen sur l’échelle d’évaluation globale par le médecin (Physician Global Assessment, PGA) et le score moyen sur l’échelle de réalisation des objectifs (Goal Attainment Scale, GAS) à la semaine 6.
Variation de la MAS et de la PGA entre le début de l’étude et les semaines 6 et 16 – 1er cycle de traitement (population ITT modifiée)
|
|
DYSPORT 2 U/kg (n=69) |
DYSPORT 8 U/kg (n=69) |
DYSPORT 16 U/kg (n=70) |
|
Changement moyen[a] du score MAS du PGMC entre le début de l’étude et Semaine 6 Semaine 16 |
-1,6 -0,9 |
-2,0* -1,2* |
-2,3*** -1,5** |
|
Changement moyen[a] du score PGA entre le début de l’étude et Semaine 6 Semaine 16 |
1,8 1,8 |
2,0 1,7 |
2,0 1,9 |
|
[a] calculé par la méthode des moindres carrés PGMC : muscles fléchisseurs du poignet ou muscles fléchisseurs du coude Pour les scores MAS et PGA, la moyenne calculée par la méthode des moindres carrés est basée sur la valeur rétrotransformée et la valeur p est basée une analyse ANCOVA / ANOVA de rang. * p≤ 0.05; **p≤ 0.001; *** p≤ 0.0001; comparativement au groupe 2 U/kg |
|||
Une amélioration de la spasticité du PGMC évaluée par l'échelle de Tardieu a été observée. Pour les fléchisseurs du coude du PGMC, l’angle de ressaut (Xv3) a été amélioré de façon statistiquement significative, à la semaine 6 pour les groupes DYSPORT traités par 8 et 16 U/kg comparativement au groupe contrôle traité par 2 U/kg, et, également à la semaine 16, pour le groupe traité par 16 U/kg. En outre, une diminution statistiquement significative du grade de spasticité (Y) aux semaines 6 et 16 par rapport aux valeurs initiales a été observée pour le groupe DYSPORT 16 U/kg par rapport au groupe contrôle traité par 2 U/kg. Dans les muscles fléchisseurs de poignet du PGMC, des améliorations statistiquement significatives de Xv3 et Y par rapport aux valeurs de base ont été observées à la semaine 6 uniquement dans le groupe DYSPORT 16 U/kg par rapport au groupe DYSPORT 2 U/kg.
Le renouvellement de l’injection a été effectué à la semaine 28 pour la majorité des sujets traités avec DYSPORT (62,3% dans le groupe DYSPORT 8 U/kg et 61,4% dans le groupe DYSPORT 16 U/kg), et seulement à la semaine 34 pour plus de 24% des patients de ces deux groupes de traitement.
5.2. Propriétés pharmacocinétiques
5.3. Données de sécurité préclinique
Administration intramusculaire (muscles striés)
Dans une étude de toxicité chronique réalisée chez des rats (jusqu'à 12 U/animal), aucune toxicité systémique n’a été observée. Les études de toxicité sur la fonction de reproduction effectuées chez des rats et des lapins en gestation, ayant respectivement reçu par injection intramusculaire quotidienne, le complexe hémagglutinine-toxine A de Clostridium botulinum à des doses de 79 U/kg et de 42 U/kg, n'ont pas entraîné de toxicité embryo-fœtale. Une toxicité maternelle sévère associée à des pertes implantatoires a été observée à des doses les plus élevées chez les deux espèces. Le complexe hémagglutinine-toxine A de Clostridium botulinum n’a démontré aucune activité tératogène chez les rats ou les lapins et aucun effet n'a été observé dans l'étude pré- et postnatale sur la génération F1 chez les rats. Chez le rat, la diminution de la fertilité observée chez le mâle et la femelle était due à la diminution du nombre d’accouplements due à la paralysie musculaire secondaire à l'administration de fortes doses.
Dans une étude de toxicité juvénile, les rats traités chaque semaine, à partir du sevrage au 21ème jour après la naissance et jusqu'à la 13ème semaine, utilisés comme modèle animal de l’enfant, âgé de 2 ans jusqu’au jeune adulte (11 administrations sur plus de 10 semaines, jusqu'à une dose totale d'environ 33 U/kg) ne présentent pas d'effets secondaires sur la croissance postnatale (y compris squelettique) et le développement des fonctions de reproduction, neurologique et neurocomportementale.
Les effets observés lors des études précliniques réalisées sur la fonction de reproduction et sur la toxicité juvénile et chronique étaient limitées à des changements locaux sur les muscles injectés liés au mécanisme d'action du complexe hémagglutinine-toxine A de Clostridium botulinum.
Administration intra-détrusorienne
Dans des études de toxicité à dose unique chez le rat et le singe, aucun effet lié à la toxine botulinique de type A n'a été retrouvé dans la vessie quelles que soient les doses testées. À des doses supérieures à la dose maximale sans effet nocif observable (NOAEL) de 67 U/kg chez le rat et de 40 U/kg chez le singe, une perte de poids corporel, une diminution de l'activité et des signes de détresse respiratoire ont été signalés chez les deux espèces. Ces signes sont révélateurs d'une toxicité systémique qui a également été observée dans des études non cliniques menées pour évaluer la sécurité de la toxine botulinique de type A administrée dans les muscles striés.
Solution d’albumine humaine à 20%, lactose monohydraté.
2 ans.
La stabilité physique et chimique en cours d’utilisation a été démontrée pendant 24 heures entre 2°C et 8°C. Du point de vue microbiologique, à moins que la méthode de reconstitution exclue le risque de contamination microbienne, le produit doit être utilisé immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l’utilisateur.
6.4. Précautions particulières de conservation
A conserver au réfrigérateur (entre + 2°C et + 8°C).
Ne pas congeler.
6.5. Nature et contenu de l'emballage extérieur
Poudre pour solution injectable en flacon (verre type I), muni d’un bouchon (halobutyl) ; boîte de 1 flacon.
6.6. Précautions particulières d’élimination et de manipulation
· Il est impératif que DYSPORT ne soit utilisé que pour le traitement d’un seul patient, au cours d’une seule séance.
· La préparation du produit doit être réalisée dans un local approprié et par du personnel expérimenté afin de minimiser le risque d’incident lors de la manipulation.
· Pour reconstituer DYSPORT 500 UNITES SPEYWOOD, utiliser une solution injectable de chlorure de sodium à 0,9 pour cent.
· Aspirer une quantité de solvant adaptée à la dilution souhaitée dans une seringue de taille adéquate.
· Après avoir nettoyé à l’alcool la partie centrale du bouchon en caoutchouc, introduire lentement ce solvant dans le flacon à température ambiante et mélanger doucement pour en dissoudre le contenu, en évitant la formation de bulles susceptibles de dénaturer le produit.
· On obtient ainsi une solution reconstituée limpide à la concentration souhaitée : EXPRIMEE EN UNITES SPEYWOOD.
· Les unités Speywood sont spécifiques de DYSPORT et ne sont pas interchangeables avec d’autres médicaments contenant de la toxine botulinique.
· Les instructions pour la reconstitution sont spécifiques à chaque dosage de DYSPORT. Les volumes de dilution correspondent à des concentrations qui sont spécifiques à chaque indication, sauf dans le cas d’incontinence urinaire chez les adultes présentant une hyperactivité neurologique du détrusor pour lequel s’appliquent des instructions spécifiques (voir ci-dessous).
|
Concentration en unités Speywood par ml (après reconstitution) |
Volume de solution injectable NaCl 0,9%* à ajouter par flacon de DYSPORT 500 Unités |
|
500 Unités/ml |
1 ml |
|
200 Unités/ml |
2,5 ml |
|
100 Unités/ml |
5 ml |
* stérile, sans conservateur à 9 mg/mL de chlorure de sodium
La préparation permet d’obtenir le volume requis de 15 mL de solution injectable reconstituée de Dysport réparti de façon égale dans deux seringues de 10 mL. Chaque seringue contient 7,5 mL de Dysport reconstitué à la même concentration.
Après la reconstitution dans la seringue, le médicament doit être utilisé immédiatement.
o Instructions de dilution pour des flacons de 500 U
§ Pour une dose de 600 U : Reconstituer deux flacons de 500 U chacun avec 2,5 mL de solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) sans conservateur. Dans la première seringue de 10 mL, prélever 1,5 mL du premier flacon et dans la deuxième seringue de 10 mL, prélever 1,5 mL du deuxième flacon. Terminer la reconstitution en ajoutant 6 mL de solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) sans conservateur dans les deux seringues et mélanger doucement.
Il est ainsi obtenu deux seringues de 10 mL, contenant chacune 7,5 mL, et correspondant à un total de 600 U de Dysport reconstitué.
§ Pour une dose de 800 U : Reconstituer deux flacons de 500 U chacun avec 2,5 mL de solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) sans conservateur. Dans la première seringue de 10 mL, prélevez 2 mL du premier flacon et dans la deuxième seringue de 10 mL, prélevez 2 mL du deuxième flacon. Terminer la reconstitution en ajoutant 5,5 mL de solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) sans conservateur dans les deux seringues et mélanger doucement.
Il est ainsi obtenu deux seringues de 10 mL, contenant chacune 7,5 mL, et correspondant à un total de 800 U de Dysport reconstitué.
o Instructions de dilution utilisant une combinaison de flacons de 500 U et 300 U (uniquement applicable pour la dose de 800 U)
§ Pour une dose de 800 U : Reconstituer le flacon de 500 U avec 2,5 mL de solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) sans conservateur et le flacon de 300 U avec 1,5 mL de solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) sans conservateur. Dans la première seringue de 10 mL, prélever 2 ml du flacon de 500 U. Dans la deuxième seringue de 10 mL, prélever les 0,5 mL restants du flacon de 500 U et la totalité des 1,5 mL du flacon de 300 U. Terminer la reconstitution en ajoutant 5,5 mL de solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) sans conservateur dans les deux seringues et mélanger doucement.
Il est ainsi obtenu deux seringues de 10 mL, contenant chacune 7,5 mL, et correspondant à un total de 800 U de Dysport reconstitué.
§
· Toute fraction de solution restante doit être éliminée de manière appropriée.
Recommandations en cas d’incident lors de la manipulation de la toxine botulinique
· La toxine est fournie et doit être utilisée à des doses thérapeutiques. Chaque ampoule correspond à une dose inférieure au 1/200e de la dose létale par voie parentérale chez l’homme. En cas d’incident lors d’une manipulation du produit qu’il soit à l’état lyophilisé ou reconstitué, les mesures appropriées décrites ci-dessous doivent être mises en route immédiatement.
· La toxine botulinique est très sensible à la chaleur et à certains agents chimiques.
· Toute projection doit être essuyée :
o soit avec un matériel absorbant imbibé d’une solution d’hypochlorite de sodium (eau de Javel) en cas de produit lyophilisé.
o soit avec un matériel absorbant sec en cas de produit reconstitué.
· Les surfaces contaminées seront nettoyées avec un matériel absorbant imbibé d’une solution d’hypochlorite de sodium (eau de Javel) puis séchées.
· En cas de bris de flacon, procéder comme indiqué ci-dessus et ramasser soigneusement les particules de verre et essuyer le produit, en évitant les coupures cutanées.
· En cas de projection cutanée, laver avec une solution d’hypochlorite de sodium (eau de Javel) puis rincer abondamment à l’eau.
· En cas de projection oculaire, rincer abondamment avec de l’eau ou avec une solution ophtalmique de rinçage oculaire.
· En cas de blessure du manipulateur (coupure, autopiqûre), procéder comme ci-dessus et prendre les mesures médicales appropriées en fonction de la dose injectée.
Recommandation pour l’élimination du matériel contaminé
· Les aiguilles, les seringues et les flacons qui ne doivent pas être vidés, seront placés après usage, dans des containers adaptés qui devront être incinérés.
· Le matériel contaminé (tissu absorbant, gants, débris d’ampoule) doit être placé dans un sac intraversable et éliminé par incinération.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
65 QUAI GEORGES GORSE
92100 BOULOGNE-BILLANCOURT
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 558 105 9 9 : Poudre pour solution injectable en flacon (verre type I), muni d’un bouchon (halobutyl) ; boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament réservé à l’usage hospitalier.
|
|
| Plan du site | Accessibilité | Contact | Téléchargement | Declaration de confidentialité | Service-Public.fr | Legifrance | Gouvernement.fr |